We assessed the safety and tolerability of ascending single doses of alirocumab in healthy Japanese subjects and evaluated the effect of alirocumab at 3 doses (50, 75, 150 mg) on low-density lipoprotein cholesterol (LDL-C) reduction in patients with primary hypercholesterolemia on atorvastatin. A randomized, single ascending-dose study of alirocumab (100, 150, 250, or 300 mg) or placebo (3:1 ratio), administered subcutaneously, was conducted in 32 healthy Japanese men. The phase 2, randomized, double-blind, placebo-controlled, parallel-group study was performed in patients with primary hypercholesterolemia (defined as calculated LDL-C ≥100 mg/dl [2.6 mmol/l]) who were on a stable dose of atorvastatin (5 to 20 mg). Patients were randomized to alirocumab (50, 75, or 150 mg) or placebo (in single 1.0-ml injection volumes) administered every 2 weeks (Q2W) for 12 weeks; the primary outcome was the mean percent change in calculated LDL-C from baseline to week 12. Single subcutaneous administration of alirocumab in healthy subjects was well tolerated over 15 weeks and resulted in highest mean percent reductions in LDL-C from baseline of approximately 40% to 60%. In the multiple-dose study, least-square mean (SE) changes in calculated LDL-C concentrations from baseline to week 12 were −54.8% (3.1%) for alirocumab 50 mg, −62.3% (3.1%) for alirocumab 75 mg, and −71.7% (3.1%) for alirocumab 150 mg, with a least-square mean (SE) difference versus placebo of −52.2% (4.3%), −59.6% (4.3%), and −69.1% (4.3%), respectively (all p <0.0001). In conclusion, alirocumab was well tolerated and significantly reduced LDL-C concentrations in Japanese patients with primary hypercholesterolemia on atorvastatin.
Highlights
- •
Alirocumab reduced low-density lipoprotein cholesterol (LDL-C) in Japanese healthy male subjects.
- •
Alirocumab reduced LDL-C by 54.8% to 71.7% in patients with hypercholesterolemia.
- •
No safety or tolerability concerns were apparent with alirocumab.
The risk of coronary artery disease and all-cause mortality rises in association with increasing concentrations of low-density lipoprotein cholesterol (LDL-C) in both Japanese and Western populations. The NIPPON DATA80 trial and the Japan Lipid Intervention Trial (J-LIT) emphasized the importance of risk factors, including dyslipidemia, hypertension, diabetes, smoking, and obesity, in relation to cardiovascular events in the Japanese population. Consequently, alongside improvements in lifestyle, the Japan Atherosclerosis Society has recommended serum LDL-C management goals, which vary according to the presence of various coronary artery disease risk factors ( Supplementary Table 1 ). A 2002 study from the Japan Lipid Assessment Program (J-LAP), involving 24,048 patients with primary hyperlipidemia on statin treatment, showed that the total cholesterol goals recommended in the Japan Atherosclerosis Society 2002 guidelines were achieved in only 53.1% of patients and the LDL-C goals in 63.4%. Furthermore, the rate of achievement was substantially higher (≥61%) in patients at low-to-moderate risk compared with those at high risk (≤45%). A more recent retrospective study, based on data collected from 2009 to 2012, indicated no improvement in achievement of lipid goals in secondary prevention, with 38% of treated Japanese patients at high cardiovascular risk failing to attain the 2012 Japan Atherosclerosis Society target of <100 mg/dl (2.6 mmol/l) for LDL-C. Alirocumab is a fully human monoclonal antibody against proprotein convertase subtilisin/kexin 9 (PCSK9). In phase 2 and 3 studies conducted primarily in Western populations, alirocumab as monotherapy or in combination with other lipid-lowering therapies (LLT) reduced LDL-C concentrations by 40% to 73%. We report here the results from initial studies of alirocumab in Japanese subjects. The principal objective of the phase 1 study was to assess the safety and tolerability of ascending single doses of alirocumab in healthy Japanese subjects. The phase 2 study was designed to assess the effect of alirocumab at 3 different doses (50, 75, and 150 mg) administered every 2 weeks (Q2W) for 12 weeks on LDL-C reduction in patients with primary hypercholesterolemia who were taking a stable dose of atorvastatin. The effect of alirocumab on other lipid parameters, as well as its safety and tolerability, was also evaluated.
Methods
We conducted 2 separate randomized placebo-controlled trials with alirocumab. The first one was a single ascending-dose study in healthy Japanese men. The second one was a multiple-dose study in patients with primary hypercholesterolemia on a background of stable atorvastatin 5 to 20 mg/day therapy. Both studies were conducted according to the Declaration of Helsinki and the International Conference on Harmonization guidelines for Good Clinical Practice. The institutional review board and ethics committee at each center approved the protocol. All participants gave written informed consent.
In the phase 1, single-dose, 15-week (106 days) study, 32 healthy men aged 20–65 years, with a body weight from 50.0 to 95.0 kg, a body mass index from 18.0 to 30.0 kg/m 2 , and serum LDL-C concentration >100 mg/dl (2.6 mmol/l) were enrolled at 1 site. None of the subjects had been taking or were currently on medications. The full entry criteria, drug administration schedule, and outcome measures are detailed in Supplementary Text 1 . In each sequential dose cohort of 100, 150, 250, and 300 mg, 8 subjects were randomly allocated (3:1) to receive a single dose of alirocumab (6 subjects) or placebo (2 subjects). Study drug was administered subcutaneously into the abdomen by research staff. Subjects were required to remain at the research facility for observation for 5 days. Blood was drawn for evaluation at baseline and at days 1, 2, 3, 4, 8, 11, 15, 22, 29, 43, 64, and 106 ( Supplementary Figure 1 ). A physical examination, assessment of vital signs, electrocardiography, blood tests, and serum antidrug antibody level measurement were also performed. Use of nonstudy drugs was not permitted during the study unless prespecified or if required, in which case the drug name, dose, duration, and indication were recorded.
The multidose study was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 2 trial. The study population comprised patients aged 20 to 75 years with primary hypercholesterolemia, defined as a calculated LDL-C concentration of ≥100 mg/dl (2.6 mmol/l), who were treated with atorvastatin at a stable dose of 5 to 20 mg (the dose range approved in Japan for primary hypercholesterolemia) for ≥6 weeks before the screening visit. The full entry criteria are detailed in Supplementary Text 2 . The study comprised a screening period with 2 options described in the following, depending on whether the patient had been on stable atorvastatin therapy for ≥6 weeks beforehand, followed by a 12-week double-blind intervention period, and an 8-week off-treatment follow-up period ( Supplementary Figure 1 ). The screening period was of 1-week duration in patients who had been receiving atorvastatin at a stable dose of 5 to 20 mg/day for ≥6 weeks before screening. Patients on LLT other than atorvastatin, who were not taking a stable dose of atorvastatin at 5 to 20 mg/day, or who were drug naïve, were asked to start atorvastatin (5 to 20 mg/day; and stop other LLT, if taking) 6 weeks before screening. The choice of atorvastatin dose in these patients was left at the investigator’s discretion. During the 12-week intervention period, patients were randomized to 1 of 3 doses of alirocumab (50, 75, or 150 mg Q2W) or placebo Q2W, all in single 1.0-ml doses administered subcutaneously by research staff into the abdomen. Randomization was stratified by the dose of atorvastatin received at randomization (<10 or ≥10 mg). Patients were instructed to remain on a stable dose of atorvastatin and on a stable diet (Japan Atherosclerosis Society Guidelines for Prevention of Atherosclerotic Cardiovascular Diseases or equivalent) throughout the study, from screening onward. The primary outcome measure was the mean percent change in calculated LDL-C from baseline to week 12, analyzed in the modified intent-to-treat population, which included all patients who underwent randomization and who had a primary end point that could be evaluated. The secondary outcome measures were the absolute change in calculated LDL-C from baseline to week 12; the proportion of patients achieving LDL-C <100 (2.6 mmol/l) or <70 mg/dl (1.8 mmol/l) at week 12; and percent and absolute changes in total cholesterol, high-density lipoprotein cholesterol (HDL-C), triglycerides, and non–HDL-C from baseline to week 12. The safety outcome measures were the occurrence of adverse events, vital signs, electrocardiographic variables, and laboratory findings (hematology, serum biochemistry), which were assessed at baseline, then every 2 weeks up to the end of the double-blind treatment period. Off-treatment follow-up visits, including safety outcome measures, were performed at weeks 16 and 20. The treatment-emergent adverse event observation period was defined as the time from the first injection of study medication to the last injection plus 70 days (10 weeks).
In the single-dose study, we estimated that 6 subjects per dose group would provide ≥80% power to detect a treatment difference of 30% (SD 15%) versus placebo in percent change from baseline in LDL-C when comparing each dose with placebo and using a 2-sided t test and significance level of 5% (not adjusted for multiple comparisons). Absolute and percent changes from baseline for each pharmacodynamic variable were analyzed using an analysis of covariance (ANCOVA) model, with the dose group as the fixed effect and the relevant baseline value as a covariate. Differences between dose groups and the placebo group, 95% confidence intervals, and p values for comparison between the treatment group and the placebo group were obtained within the framework of ANCOVA.
In the multiple-dose study, we estimated that 25 patients per arm would have a >99% power to detect a 30% (SD 20%) difference in LDL-C percent change from baseline to week 12 between alirocumab and placebo, assuming a 5% rate of nonevaluable primary end points and using a 2-sided t test at the 0.05 significance level. A hierarchical testing procedure was used to address multiplicity of pairwise comparisons versus placebo. The hierarchical testing sequence was continued only when the higher order test was statistically significant at the 5% level to ensure that overall type I error was controlled at the 5% level. The primary efficacy analysis was conducted in the modified intent-to-treat population, defined as the randomized population with an evaluable primary end point. The efficacy period was the time from the first injection of study drug up to 21 days after the last injection. Patients in the modified intent-to-treat population were analyzed according to the treatment group allocated by randomization. Percent change in lipid variables from baseline to week 12 was analyzed using an ANCOVA model, with treatment group and randomization strata of atorvastatin dose (<10, ≥10 mg) as fixed effects and baseline LDL-C value as covariate. Throughout, each dose of alirocumab was compared with placebo using appropriate contrasts and hierarchic procedure, and the 95% adverse event of the difference versus placebo (not adjusted for multiple comparisons) was provided. In case of missing week-12 LDL-C on-treatment data, the last observation-carried-forward principle was used. The proportions of patients who achieved LDL-C values <100 (2.6 mmol/l) or <70 mg/dl (1.8 mmol/l) at week 12 were analyzed using a stratified exact conditional logistic regression model with treatment group and baseline LDL-C level as effects and atorvastatin dose (<10, ≥10 mg) as stratum. The safety population was defined as the randomized population who received at least 1 dose or partial dose of study treatment. The treatment-emergent adverse event period was defined as the time from the first dose of study drug up to the last dose plus 70 days.
Results
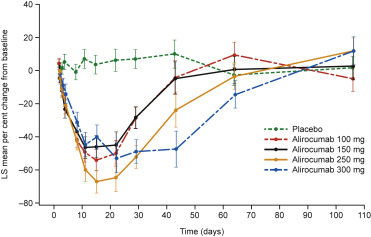
Thirty-two healthy men were randomized to alirocumab or placebo in the single-dose study ( Supplementary Figure 2 ). Their mean (SD) baseline calculated LDL-C values ranged from 105.3 (12.0) to 130.8 (15.3) mg/dl ( Supplementary Table 2 ). Single subcutaneous administration of alirocumab in healthy subjects resulted in a significant and sustained mean LDL-C reduction from baseline. The mean percent reductions in LDL-C from baseline were from 40% to 60% and were achieved from day 11 until day 22 after treatment for the 100-, 150-, and 250-mg doses and from day 11 until day 43 for the 300-mg dose ( Figure 1 ). No deaths, serious adverse events, or other significant events were reported. Orthostatic hypotension developed 30 minutes after drug administration in a 26-year-old man in the alirocumab 250-mg dose group; his systolic and diastolic values returned to normal within 30 minutes. A 39-year-old man in the placebo group reported fatigue. Both adverse events were judged to be unrelated to the study drug and were of mild intensity, and the subjects fully recovered. No subjects had injection-site reactions. Few potentially clinically significant abnormalities in vital signs, laboratory and electrocardiographic variables were reported, with no dose–incidence relation, and were not considered to be clinically significant ( Supplementary Tables 3 and 4 ). The incidences and titers of antidrug antibody were similar across alirocumab dose groups ( Supplementary Table 5 ). Serum alirocumab concentrations were similar in antidrug antibody-positive and -negative subjects across alirocumab dose groups ( Supplementary Figure 3 ).
We screened 162 patients in the multiple-dose study and 100 were randomized to alirocumab or placebo ( Supplementary Figure 4 ). The patients’ characteristics were broadly similar across the 4 groups ( Table 1 ). For the primary modified intent-to-treat efficacy analysis, least-square mean (SE) reductions in calculated LDL-C concentrations from baseline to week 12 were statistically significant at all doses (all p <0.0001; Table 2 ). A substantial dose-dependent reduction was achieved in calculated LDL-C in the first 2 weeks in the alirocumab groups, which was sustained through the 12-week treatment period ( Figure 2 ). All the patients in the 3 alirocumab groups achieved LDL-C <100 mg/dl (2.6 mmol/l) at week 12, versus 2 (8.0%) patients in the placebo group. More than 80% patients in each alirocumab group (n = 21, 84.0% in both the 50- and 75-mg groups; n = 22, 88.0% in the 150-mg group) achieved LDL-C <70 mg/dl (1.8 mmol/l), versus none in the placebo group. Alirocumab demonstrated a dose-dependent increase in the number of patients who achieved a ≥50% reduction in LDL-C (n = 15, 60.0%; n = 20, 80.0%; n = 23, 92.0%, respectively), versus none of the patients in the placebo group. The numbers of patients who displayed at least 1 LDL-C value of <25 mg/dl (0.7 mmol/l) during the efficacy period were 6 (24.0%) for alirocumab 50 mg, 10 (40.0%) for 75 mg, and 14 (56.0%) for 150 mg. Alirocumab also resulted in statistically significant decreases in total cholesterol, non–HDL-C, apolipoprotein B (p <0.0001 at all concentrations), and lipoprotein (a) (all p <0.01). Triglyceride concentrations were statistically significantly reduced with the 75- and 150-mg doses of alirocumab (p ≤0.02). Treatment-emergent adverse events were reported in 52.0% of patients on alirocumab 50 mg, 48.0% on alirocumab 75 mg, 64.0% on alirocumab 150 mg, versus 32.0% of patients on placebo ( Table 3 ). Two patients had serious adverse events: 1 patient in the alirocumab 150-mg group was diagnosed with breast cancer and discontinued study treatment prematurely and 1 patient in the placebo group experienced severe vertigo. Neither event was judged to be related to the study medication. No deaths were reported during the study. Injection-site reactions were reported in 7 (9.3%) patients on alirocumab and 1 (4.0%) patient on placebo. The reaction was assessed as related to the study medication in 1 patient on alirocumab 150 mg, leading to study drug discontinuation, and of mild intensity in the remaining 7 patients. One patient, on alirocumab 50 mg, experienced an increase >3 times the upper limit of normal (ULN) in both alanine aminotransferase and aspartate aminotransferase. One patient on alirocumab 75 mg had an increase in alanine phosphatase >1.5 times the ULN, and 3 patients (1 in each alirocumab arm) had creatinine phosphokinase elevation >3 (<10) times the ULN. None of the patients experienced a total bilirubin value >1.5 times the ULN. Fifteen patients, all in the alirocumab groups, had 2 consecutive LDL-C values <25 mg/dl (0.7 mmol/l); of these patients, 6 had 2 consecutive LDL-C values <15 mg/dl (0.4 mmol/l). Among the patients with 2 consecutive LDL-C values <25 mg/dl, adverse events were reported in 1 of 2 on alirocumab 50-mg group, 2 of 4 on alirocumab 75 mg, and 5 of 9 on alirocumab 150 mg, with no serious adverse events or discontinuations of study treatment. The rates of adverse events in this group were no different from those in the overall treated population ( Table 3 ). Additional safety data (vital signs and electrocardiographic variables) are detailed in Supplementary Table 6 .
| Variable | Placebo (n = 25) | Alirocumab 50 mg Q2W (n = 25) | Alirocumab 75 mg Q2W (n = 25) | Alirocumab 150 mg Q2W (n = 25) |
|---|---|---|---|---|
| Age (years) | 58.6 (9.2) | 57.8 (12.3) | 56.3 (12.0) | 58.2 (8.8) |
| Women | 11 (44%) | 16 (64%) | 12 (48%) | 16 (64%) |
| Body mass index (kg/m 2 ) ∗ | 25.8 (3.2) | 24.3 (3.9) | 24.5 (3.3) | 24.2 (4.4) |
| Any medical history † | 24 (96%) | 25 (100%) | 23 (92%) | 23 (92%) |
| Hypertension | 13 (52%) | 9 (36%) | 8 (32%) | 5 (20%) |
| Diabetes mellitus (type 2) | 4 (16%) | 5 (20%) | 2 (8%) | 5 (20%) |
| Cerebrovascular disease | 2 (8%) | 2 (8%) | 1 (4%) | 0 |
| Peripheral artery disease | 1 (4%) | 1 (4%) | 0 | 0 |
| Coronary artery disease | 0 (0%) | 1 (4%) | 0 | 0 |
| Lipid parameter | ||||
| Low-density lipoprotein cholesterol (Friedewald formula) (mg/dL) | 121.0 (21.1) | 122.2 (16.6) | 120.9 (16.7) | 120.5 (16.2) |
| Range (minimum : maximum) | 89 : 184 | 95 : 151 | 98 : 169 | 93 : 148 |
| Total cholesterol (mg/dL) | 203.6 (27.7) | 205.2 (19.9) | 209.2 (24.3) | 212.5 (22.7) |
| High-density lipoprotein cholesterol (mg/dL) | 55.3 (11.2) | 58.8 (12.4) | 61.1 (15.3) | 68.1 (13.5) |
| Triglycerides, median (quartile 1, quartile 3) (mg/dL) | 128.0 (101.5, 156.5) | 126.0 (90.5, 136.5) | 119.5 (86.5, 182.5) | 107.0 (87.0, 124.5) |
| Non–high-density lipoprotein cholesterol (mg/dL) | 148.2 (27.0) | 146.4 (18.1) | 148.0 (23.2) | 144.4 (20.7) |
| Apolipoprotein B (g/L) | 1.0 (0.2) | 1.0 (0.1) | 1.0 (0.2) | 1.0 (0.2) |
| Apolipoprotein A1 (g/L) | 1.5 (0.2) | 1.5 (0.2) | 1.6 (0.3) | 1.7 (0.3) |
| Apolipoprotein B:A1 | 0.7 (0.2) | 0.6 (0.1) | 0.6 (0.2) | 0.6 (0.1) |
| Lipoprotein (a), median (quartile 1, quartile 3) (mg/dL) | 11.7 (6.9, 26.8) | 9.6 (6.7, 23.4) | 14.8 (7.5, 28.9) | 16.9 (7.3, 39.5) |
| Other laboratory variables | ||||
| Glycated hemoglobin (%) | 6.1 (0.6) | 6.0 (0.8) | 5.8 (0.7) | 5.9 (0.6) |
| Glycated hemoglobin | ||||
| <5.7% | 7 (28%) | 9 (36%) | 13 (52%) | 9 (36%) |
| 5.7% to <6.5% | 11 (44%) | 11 (44%) | 9 (36%) | 11 (44%) |
| ≥6.5% | 7 (28%) | 5 (20%) | 3 (12%) | 5 (20%) |
| Plasma glucose, fasting (mg/dL) | 109.2 (17.8) | 108.8 (20.4) | 100.5 (14.6) | 104.9 (16.0) |
| High-sensitivity C-reactive protein (mg/L) | 0.8 (0.6) | 1.1 (2.5) | 1.3 (2.5) | 0.5 (0.6) |
∗ Body mass index is the weight in kg divided by the square of the height in m.
† Medical/surgical history other than disease being treated in the study. Patients can be counted in several categories.
| Variable | Placebo (n = 25) | Alirocumab | ||
|---|---|---|---|---|
| 50 mg Q2W (n = 25) | 75 mg Q2W (n = 25) | 150 mg Q2W (n = 25) | ||
| Primary endpoint: | ||||
| Low-density lipoprotein cholesterol (calculated) (modified intention to treat) | ||||
| Week 12, mean (SD) (mg/dL) ∗ | 116.0 (16.8) | 54.4 (16.3) | 46.4 (18.5) | 35.0 (24.1) |
| LS mean (SE) change from baseline (%) | –2.7 (3.1) | –54.8 (3.1) | –62.3 (3.1) | –71.7 (3.1) |
| LS mean (SE) difference vs. placebo (%) | – | –52.2 (4.3) | –59.6 (4.3) | –69.1 (4.3) |
| P-value vs. placebo | – | <0.0001 | <0.0001 | <0.0001 |
| Secondary endpoints | ||||
| LS mean (SE) absolute difference in low-density lipoprotein cholesterol vs. placebo (mg/dL) | – | –62.1 (5.0) | –69.5 (5.0) | –85.8 (3.6) |
| P-value vs. placebo | <0.0001 | <0.0001 | <0.0001 | |
| Total cholesterol | ||||
| Week 12, mean (SD) (mg/dL) ∗ | 200.5 (20.5) | 139.7 (22.2) | 133.2 (23.3) | 123.7 (28.3) |
| Mean (SD) change from baseline (%) | –0.6 (11.3) | –31.5 (11.8) | –36.4 (7.9) | –41.8 (11.4) |
| LS mean (SE) change from baseline (%) | –1.2 (2.1) | –31.9 (2.1) | –36.3 (2.1) | –41.4 (2.1) |
| LS mean (SE) difference vs. placebo (%) | – | –30.7 (3.0) | –35.1 (3.0) | –40.2 (3.0) |
| P-value vs. placebo | – | <0.0001 | <0.0001 | <0.0001 |
| Absolute change from baseline, mean (SD) (mg/dL) | –3.1 (24.6) | –65.5 (26.9) | –76.0 (18.5) | –88.8 (25.7) |
| High-density lipoprotein cholesterol | ||||
| Week 12, mean (SD) (mg/dL) ∗ | 56.4 (12.4) | 62.3 (13.5) | 63.9 (15.0) | 68.7 (12.5) |
| Mean (SD) change from baseline (%) | 2.1 (8.0) | 6.4 (8.6) | 5.7 (12.6) | 2.0 (12.5) |
| LS mean (SE) difference vs. placebo (%) | – | 5.3 (2.8) | 5.2 (2.8) | 3.4 (3.0) |
| P-value vs. placebo | – | 0.06 | 0.07 | 0.25 |
| Absolute change from baseline, mean (SD) (mg/dL) | 1.1 (4.6) | 3.6 (5.2) | 2.8 (5.9) | 0.6 (8.3) |
| Triglycerides | ||||
| Week 12, median (quartile 1, quartile 3) (mg/dL) ∗ | 136.0 (105.0, 169.0) | 97.0 (70.0, 139.0) | 94.0 (75.0, 152.0) | 98.0 (72.0, 115.0) |
| Median (quartile 1, quartile 3), change from baseline (%) | 1.3 (–13.1, 22.5) | –21.1 (–28.2, 4.2) | –10.7 (–25.0, 6.5) | –15.0 (–24.1, 0.0) |
| Effect size estimate (95% CI) | – | –17.2 (–29.4 to 2.7) | –16.9 (–34.7 to –0.4) | –19.4 (–37.5 to –4.7) |
| P-value vs. placebo | – | 0.06 | 0.02 | 0.006 |
| Absolute change from baseline, median (quartile 1, quartile 3) (mg/dL) | 2.0 (–14.5, 28.5) | –19.0 (–32.0, 4.0) | –10.0 (–24.5, 4.5) | –16.0 (–40.0, 0.0) |
| Non–high-density lipoprotein cholesterol | ||||
| Week 12, mean (SD) (mg/dL) ∗ | 144.2 (20.1) | 77.5 (22.0) | 69.3 (21.5) | 54.9 (23.1) |
| Mean (SD) change from baseline (%) | –1.2 (15.3) | –46.4 (16.5) | –53.5 (10.5) | –62.1 (14.4) |
| LS mean (SE) change from baseline (%) | –0.9 (2.9) | –46.3 (2.9) | –53.1 (2.9) | –62.3 (2.9) |
| LS mean (SE) difference vs. placebo (%) | – | –45.4 (4.0) | –52.3 (4.0) | –61.4 (4.0) |
| P-value vs. placebo | – | <0.0001 | <0.0001 | <0.0001 |
| Absolute change from baseline, mean (SD) (mg/dL) | –4.0 (24.1) | –68.9 (27.4) | –78.7 (18.2) | –89.5 (24.3) |
| Apolipoprotein B | ||||
| Week 12, mean (SD) (g/L) ∗ | 1.0 (0.1) | 0.5 (0.2) | 0.5 (0.2) | 0.4 (0.2) |
| Percent change from baseline, mean (SD) | –2.8 (15.1) | –43.7 (19.9) | –48.9 (12.2) | –60.2 (16.5) |
| LS mean (SE) change from baseline (%) | –2.3 (3.3) | –43.5 (3.3) | –48.6 (3.3) | –60.0 (3.3) |
| LS mean (SE) difference vs. placebo (%) | – | –41.2 (4.7) | –46.3 (4.6) | –57.7 (4.6) |
| P-value vs. placebo | – | <0.0001 | <0.0001 | <0.0001 |
| Apolipoprotein A-1 | ||||
| Week 12, mean (SD) (g/L) ∗ | 1.5 (0.2) | 1.6 (0.2) | 1.7 (0.2) | 1.7 (0.2) |
| Mean (SD) change from baseline (%) | –1.5 (7.9) | 6.1 (6.5) | 4.9 (9.7) | 3.5 (9.7) |
| LS mean (SE) difference vs. placebo (%) | – | 8.1 (2.1) | 7.8 (2.1) | 7.9 (2.2) |
| P-value vs. placebo | – | 0.0002 | 0.0003 | 0.0004 |
| Lipoprotein (a) | ||||
| Week 12, median (quartile 1, quartile 3) (mg/dL) ∗ | 13.6 (4.9, 23.1) | 7.3 (2.0, 14.8) | 7.1 (4.0, 15.2) | 10.0 (2.0, 23.9) |
| Median (quartile 1, quartile 3), change from baseline (%) | –3.7 (–26.2, 8.4) | –35.6 (–52.5, –7.5) | –40.2 (–62.9, –27.4) | –43.3 (–69.4, –14.2) |
| Effect size estimate (95% CI) | – | –28.1 (–46.5, –5.5) | –37.5 (–55.0, –18.0) | –34.1 (–59.1, –8.6) |
| P-value vs. placebo | – | 0.0064 | 0.0003 | 0.0009 |
| Apolipoprotein B/A-1 ratio | ||||
| Week 12, mean (SD) ∗ | 0.7 (0.2) | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) |
| LS mean (SE) difference vs. placebo, absolute change | – | –0.3 (0.03) | –0.3 (0.03) | –0.4 (0.03) |
| P-value vs. placebo | – | <0.0001 | <0.0001 | <0.0001 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


