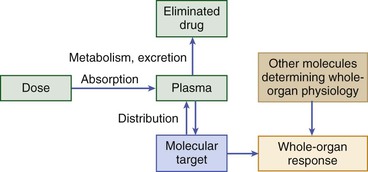
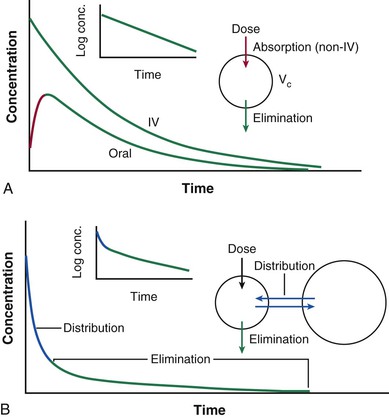
Dan M. Roden Drug treatment makes up a large fraction of total health care costs. In 2008, the total cost of healthcare in the United States was approximately $2.5 trillion, and more than 10% was spent on prescription drugs.1 Cardiovascular disease makes up the largest subcategory in this spending: The American Heart Association estimated that the 2008 cost of care for cardiovascular disease was $298 billion, and total prescription drug costs for cardiovascular care were $33 billion.2 Patients vary in their responses to drug treatment, and multiple mechanisms can be invoked, such as poor compliance, variable impact of diverse disease mechanisms on drug actions, drug interactions, and an increasingly recognized role of genomic variation. Indeed, adverse drug reactions across all therapeutic categories are estimated to be the fourth to the sixth most common cause of death in the United States, costing $19 to $27 billion annually, and accounting directly for 3% to 6% of all hospital admissions.3 This chapter outlines principles of drug action, the major mechanisms underlying variability in drug effects, and current and future approaches to enable the safest and most effective therapy for an individual patient. The fundamental assumption underlying administration of any drug is that the real or expected benefit exceeds the anticipated risk. The benefits of drug therapy are initially defined in small clinical trials, perhaps involving several thousand patients, before a drug’s marketing and approval. Ultimately, the efficacy and safety profiles of any drug are determined after the compound has been marketed and used widely in hundreds of thousands of patients. When a drug is administered for the acute correction of a life-threatening condition, the benefits are often self-evident; insulin for diabetic ketoacidosis, nitroprusside for hypertensive encephalopathy, and lidocaine for ventricular tachycardia are examples. Extrapolation of such immediately obvious benefits to other clinical situations may not be warranted, however. The efficacy of lidocaine to terminate ventricular tachycardia led to its widespread use as a prophylactic agent in cases of acute myocardial infarction, until it was recognized that in this setting, the drug does not alter mortality rates. The outcome of the Cardiac Arrhythmia Suppression Trial (CAST) highlights the difficulties in extrapolating from an incomplete understanding of physiology to chronic drug therapy. CAST tested the hypothesis that suppression of ventricular ectopic activity, a recognized risk factor for sudden death after myocardial infarction, would reduce mortality; this notion was highly ingrained in cardiovascular practice in the 1970s and 1980s. In CAST, sodium channel–blocking antiarrhythmics did suppress ventricular ectopic beats but also unexpectedly increased mortality threefold. Similarly, with the development of a first-generation cholesterol ester transport protein (CETP) inhibitor, the goal of elevation of high-density lipoprotein (HDL) levels was achieved, but with an accompanying increase in mortality. Thus, the use of arrhythmia suppression or of HDL elevation as a surrogate marker did not produce the desired drug action, reduction in mortality, probably because the underlying pathophysiology or full range of drug actions were incompletely understood. Similarly, drugs with positive inotropic activity augment cardiac output in patients with heart failure but also are associated with an increase in mortality, probably as a consequence of drug-induced arrhythmias. Nevertheless, clinical trials with these agents suggest symptom relief. Thus, the prescriber and the patient may elect therapy with positive inotropic drugs to realize this benefit while recognizing the risk. This complex decision making is at the heart of the broad concept of personalized medicine that incorporates into the care of an individual patient not only genomic (or other) markers of variable drug responses but also factors such as patients’ understanding of their disease and their willingness to tolerate minor or serious risks of treatment. The risks of drug therapy may be a direct extension of the pharmacologic actions for which the drug is actually being prescribed. Excessive hypotension in a patient taking an antihypertensive agent and bleeding in a patient taking a platelet IIb/IIIa receptor antagonist are examples. In other cases, adverse effects develop as a consequence of pharmacologic actions that were not appreciated during a drug’s initial development and use in patients. Rhabdomyolysis occurring with HMG-CoA reductase inhibitors (statins), angioedema developing during ACE inhibitor therapy, and torsades de pointes arising during treatment with noncardiovascular drugs such as thioridazine or pentamidine are examples. Of importance, these rarer but serious effects generally become evident only after a drug has been marketed and extensively used. Even rare adverse effects can alter the overall perception of risk versus benefit and can prompt removal of the drug from the market, particularly if alternate therapies thought to be safer are available. For example, withdrawal of the first insulin sensitizer, troglitazone, after recognition of hepatotoxicity was further spurred by the availability of other new drugs in this class. The recognition of multiple cyclooxygenase (COX) isoforms led to the development of specific COX-2 inhibitors to retain aspirin’s analgesic effects but reduce gastrointestinal side effects. However, one of these, rofecoxib, was withdrawn because of an apparent increase in cardiovascular mortality. The events surrounding the withdrawal of rofecoxib have important implications for drug development and utilization. First, specificity achieved by targeting a single molecular entity may not necessarily reduce adverse effects; one possibility is that by inhibiting COX-2, the drug removes a vascular protective effect of prostacyclin. Second, drug side effects may include not only readily identifiable events such as rhabdomyolysis or torsades de pointes but also an increase that may be difficult to detect in events such as myocardial infarction that are common in the general population. Two major processes determine how the interaction between a drug and its target molecule(s) can generate variable drug actions in a patient (Fig. 9-1). The first, pharmacokinetics, describes drug delivery to and removal from the target molecule and includes the processes of absorption, distribution, metabolism, and excretion—collectively termed drug disposition. The second process, pharmacodynamics, describes how the interaction between a drug and its molecular target(s) generates downstream molecular, cellular, whole-organ, and whole-body effects. The framework shown in Figure 9-1 identifies a series of genes that mediate clinical drug actions, and in which variants may thus contribute to variable drug actions. These genes encode drug-metabolizing enzymes, drug transport molecules, drug targets, and molecules modulating the biology in which the drug-target interaction occurs. The latter include molecular perturbations that cause the disease being targeted. Pharmacogenetics describes the concept that individual variants in the genes controlling these processes contribute to variable drug actions, whereas pharmacogenomics describes the way in which variability across multiple genes, up to whole genomes, explains differences in drug response among individuals and populations. Presented next is an overview of broad principles of pharmacokinetics, pharmacodynamics, and pharmacogenomics, followed by more detailed discussions of the specific genes, their function, and important variants influencing cardiovascular drug responses. Pharmacokinetic Principles Administration of an intravenous drug bolus results in maximal drug concentrations at the end of delivery of the bolus, followed by a decline in plasma drug concentrations over time (Fig. 9-2A), generally due to drug elimination. The simplest case is one in which this decline occurs monoexponentially over time. A useful parameter to describe this decline is the half-life (t1/2), the time in which 50% of the drug is eliminated; for example, after two half-lives, 75% of the drug has been eliminated, after three half-lives, 87.5%. A monoexponential process can be considered almost complete in four or five half-lives. In some cases, the decline of drug concentrations following administration of an intravenous bolus dose is multiexponential. The most common explanation is that drug is not only eliminated (represented by the terminal portion of the time-concentration plot) but also undergoes more rapid distribution to peripheral tissues. Just as elimination may be usefully described by a half-life, distribution half-lives also can be derived from curves such as those shown in Figure 9-2B. The plasma concentration measured immediately after a bolus dose can be used to derive a volume into which the drug is distributed. When the decline of plasma concentrations is multiexponential, multiple distribution compartments can be defined; these volumes of distribution can be useful in considering dose adjustments in cases of disease but rarely correspond exactly to any physical volume, such as plasma or total body water. With drugs that are highly tissue-bound (e.g., some antidepressants), the volume of distribution can exceed total body volume by orders of magnitude. Drugs are often administered by nonintravenous routes, such as oral, sublingual, transcutaneous, or intramuscular. Such routes of administration differ from the intravenous route in two ways (see Fig. 9-2A). First, concentrations in plasma demonstrate a distinct rising phase as the drug slowly enters plasma. Second, the total amount of drug that actually enters the systemic circulation may be less than that achieved by the intravenous route. The relative amount of drug entering by any route, compared with the same dose administered intravenously, is termed bioavailability. Some drugs undergo such extensive presystemic metabolism that the amount of drug required to achieve a therapeutic effect is much greater (and often more variable) than that required for the same drug administered intravenously. Thus, small doses of intravenous propranolol (5 mg) may achieve heart rate slowing equivalent to that observed with much larger oral doses (80 to 120 mg). Propranolol is actually well absorbed but undergoes extensive metabolism in the intestine and liver before entering the systemic circulation. Another example is that of amiodarone; its physicochemical characteristics make it only 30% to 50% bioavailable when administered orally. Thus, an intravenous infusion of 0.5 mg/min (720 mg/day) is equivalent to 1.5 to 2 g/day orally. Drug elimination occurs by metabolism followed by the excretion of metabolites and unmetabolized parent drug, generally by the biliary tract or kidneys. This process can be quantified as clearance, the volume that is cleared of drug in any given period. Clearance may be organ-specific (e.g., renal clearance, hepatic clearance) or whole-body clearance. Drug metabolism is conventionally divided into phase I oxidation and phase II conjugation, both of which enhance water solubility and, consequently, biliary or renal elimination. The most common enzyme systems mediating phase I drug metabolism are those of the cytochrome P-450 superfamily, termed CYPs. Multiple CYPs are expressed in human liver and other tissues. A major source of variability in drug action is variability in CYP expression and/or genetic variants that alter CYP activity. Table 9-1 lists CYPs and other drug-metabolizing enzymes important in cardiovascular therapy. Excretion of drugs or their metabolites into the urine or bile is accomplished by glomerular filtration or specific drug transport molecules, whose level of expression and genetic variation are only now being explored. One widely studied transporter is P-glycoprotein, the product of expression of the MDR1 (or ABCB1) gene. Originally identified as a factor mediating multiple drug resistance in patients with cancer, P-glycoprotein expression is now well recognized in normal enterocytes, hepatocytes, renal tubular cells, the endothelium of the capillaries forming the blood-brain barrier, and the testes. In each of these sites, P-glycoprotein expression is restricted to the apical aspect of polarized cells, where it acts to enhance drug efflux. In the intestine, P-glycoprotein pumps substrates back into the lumen, thereby limiting bioavailability. In the liver and kidney, it promotes drug excretion into bile or urine. In central nervous system capillary endothelium, P-glycoprotein–mediated efflux is an important mechanism limiting drug access to the brain. Drug transporters play a role not only in drug elimination but also in drug uptake into many cells, including hepatocytes and enterocytes. TABLE 9-1 Proteins Important in Drug Metabolism and Elimination * Clinically important genetic variants described; see text. † Prodrug bioactivated by drug metabolism. Full CYP listing is available at http://medicine.iupui.edu/flockhart. Drugs can exert variable effects, even in the absence of pharmacokinetic variability. As indicated in Figure 9-1, this can arise as a function of variability in the molecular targets with which drugs interact to achieve their beneficial and adverse effects, as well as variability in the broader biologic context within which the drug-target interaction takes place. Variability in the number or function of a drug’s target molecules can arise because of genetic factors (see later) or because disease alters the number of target molecules or their state (e.g., changes in the extent of phosphorylation). Simple examples of variability in the biologic context are high dietary salt, which can inhibit the antihypertensive action of beta blockers, and hypokalemia, which increases the risk for drug-induced QT prolongation. In addition, disease itself can modulate drug response. For example, the effect of lytic therapy in a patient with no clot is manifestly different from that in a patient with acute coronary thrombosis, or the vasodilating effects of nitrates, beneficial in patients with coronary disease with angina, can be catastrophic in patients with aortic stenosis. These examples highlight the requirement for precision in diagnosis to avoid situations in which risk outweighs potential benefit. One hope is that emerging genomic or other molecular approaches can add to this precision. The targets with which drugs interact to produce beneficial effects may or may not be the same as those with which drugs interact to produce adverse effects. Drug targets may be in the circulation, at the cell surface, or within cells. Many newer drugs have been developed to interact with a specific drug target; examples of such targets are 3-hydroxy-3-methyl-glutaryl–coenzyme A (HMG-CoA) reductase, angiotensin-converting enzyme (ACE), G protein–coupled receptors (e.g., alpha, beta, angiotensin II, histamine), and platelet IIb/IIIa receptors. Such targets generally are identified in the course of basic mechanistic studies; a very appealing newer approach is to use modern genetic techniques to identify DNA variants associated with desired phenotypes, such as absence of myocardial infarction, as a clue to identify new drug targets.4 On the other hand, many drugs widely used in cardiovascular therapeutics were developed when the technology to identify specific molecular targets simply was not available; digoxin, amiodarone, and aspirin are examples. Some, like amiodarone, have many drug targets. In other cases, however, even older drugs turn out to have rather specific molecular targets. The actions of digitalis glycosides are mediated primarily by the inhibition of Na+,K+-ATPase. Aspirin permanently acetylates a specific serine residue on the COX enzyme, an effect that is thought to mediate its analgesic effects and its gastrointestinal toxicity. With repeated doses, drug levels accumulate to a steady state, the condition under which the rate of drug administration is equal to the rate of drug elimination in any given period. Drug accumulation to steady state is near-complete in four to five elimination half-lives (see Fig. 9-3). For many drugs, the target molecule is in or readily accessible from plasma, so this time course also describes the development of pharmacologic effects. However, in other cases, whereas steady-state plasma concentrations are achieved in four to five elimination half-lives, steady-state drug effects take longer to achieve and several explanations are possible. First, an active metabolite may need to be generated to achieve drug effects. Second, time may be required for translation of the drug effect at the molecular site to a physiologic endpoint; inhibition of synthesis of vitamin K–dependent clotting factors by warfarin ultimately leads to a desired elevation of the international normalized ratio (INR), but the development of this desired effect occurs only as levels of clotting factors fall. Third, penetration of a drug into intracellular or other tissue sites of action may be required before development of drug effect. One mechanism underlying such penetration is the variable function of specific drug uptake and efflux transport proteins that control intracellular drug concentrations. Pharmacogenomic Principles
Drug Therapeutics and Personalized Medicine
Importance of Correct Drug Use
The Key Decision in Drug Therapy: Risk Versus Benefit
Mechanisms Underlying Variability in Drug Action
PROTEIN
SUBSTRATES
CYP3A4, CYP3A5
Erythromycin, clarithromycin; quinidine, mexiletine; many benzodiazepines; cyclosporine, tacrolimus; many antiretrovirals;
HMG CoA reductase inhibitors (atorvastatin, simvastatin, lovastatin; not pravastatin); many calcium channel blockers
CYP2D6*
Some beta blockers—propranolol, timolol, metoprolol, carvedilol
Propafenone; desipramine and other tricyclics; codeine†; tamoxifen†; dextromethorphan
CYP2C9*
Warfarin, phenytoin, tolbutamide, losartan†
CYP2C19*
Omeprazole, clopidogrel†
P-glycoprotein
Digoxin
N-acetyl transferase*
Procainamide, hydralazine, isoniazid
Thiopurine methyltransferase*
6-Mercaptopurine, azathioprine
Pseudocholinesterase*
Succinylcholine
UDP-glucuronosyltransferase*
Irinotecan†
SLCO1B1*
Simvastatin and other statins; methotrexate; troglitazone; bosentan
Pharmacodynamic Principles
Time Course of Drug Effects
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Drug Therapeutics and Personalized Medicine
9