Double-Outlet Right Ventricle
Gail E. Wright
Katsuhide Maeda
Norman H. Silverman
Frank L. Hanley
Stephen J. Roth
Definition
Double-outlet right ventricle (DORV) is a type of ventriculoarterial connection in which both great arteries arise 50% or more from the right ventricle (RV). This chapter focuses on DORV with concordant atrioventricular (AV) connection with two ventricles. DORV may occur in univentricular hearts, particularly in the constellation of heterotaxy syndrome, and with AV discordance. Those DORV variants are described in other chapters (see Chapters 50, 51).
Although there were reports of hearts with DORV anatomy as early as 1893, the term double-outlet right ventricle came into use in 1957 at the time of the first surgical repair of a patient with this lesion at the Mayo Clinic (1,2). Prior descriptions, including the original report in 1949 of what is now known as Taussig–Bing heart, included DORV variants as a form of partial transposition of the great arteries (3,4,5). There has been much debate about this terminology. For the purpose of this chapter, DORV is considered within the spectrum of conotruncal defects, with some forms exhibiting transposition physiology in which the pulmonary arterial oxygen saturation is higher than the aortic oxygen saturation. However, DORV and transposition are considered distinct anatomically, because discordant ventriculoarterial connections are core to the anatomic definition of transposition. In 1961, Neufeld et al. developed a classification of DORV with and without pulmonic stenosis (PS) (6,7,8). Subsequently in 1972, Lev et al. categorized many types of DORV (9). Shortly thereafter, Sridaromont et al. reported on hemodynamic and angiocardiographic correlations of anatomic variants (10,11). DORV nomenclature was reviewed more recently in the Congenital Heart Surgery Nomenclature and Database Project (12,13).
Incidence
DORV accounts for fewer than 1% of all congenital heart defects. Its incidence is approximately 0.06 cases per 1,000 live births (14). There is no known racial or gender predilection, and no associated genetic defect has been identified.
Morphology
DORV falls within the morphologic spectrum of conotruncal defects. As with much of congenital heart disease, the physiology and treatment of these defects are derived from the embryology and morphology.
With the exception of truncus arteriosus, which occurs due to failure of septation, other conotruncal defects are essentially rotational defects. The development of the conal septum is thought to drive that rotation. The variability among the individual lesions is best understood in terms of the spectrum of development of the conal septum, which determines the relative position of the two semilunar valves to the ventricles. The more conal muscle present beneath a semilunar valve, the more that valve is pushed superiorly and anteriorly, and the more likely it is that the associated great artery will align with the RV (15,16).
In the normal heart, the pulmonary valve sits up on the conus, a circular tube of muscle, and is positioned anteriorly and superiorly (17). In contrast, the aortic, mitral, and tricuspid valves are all attached to the central fibrous body of the heart. The conal muscle beneath the aortic valve largely resorbs, leaving the aorta positioned inferiorly and posteriorly (Fig. 49.1).
In conotruncal defects, there is a spectrum between hearts in which no conus exists beneath the aorta, as seen in tetralogy of Fallot, and no conus exists under the pulmonary valve, as with transposition of the great arteries (Fig. 49.1). At each end of this spectrum, it is “all or nothing” in terms of the conus. DORV falls in the middle of the spectrum, with forms that have variable amounts of conus under each semilunar valve scattered across the continuum (18).
One common form of DORV is anatomically and physiologically similar to tetralogy of Fallot (19). There is a near-normal length of conus beneath the pulmonary valve and minimal conus beneath the aortic valve. Consequently, there is no aorto-mitral continuity, and the pulmonary valve is anterior and superior. At the other end of the spectrum, another DORV variant has conus mostly under the aortic valve but has a small amount of conus under the pulmonary valve, resulting in the loss of pulmonary-mitral continuity. This form is physiologically similar to transposition of the great arteries. In the middle is a type which has equal bilateral conus, such that the great arteries are side by side, with neither vessel tucked in posteriorly. Between the tetralogy type and the transposition type, there are many “shades of gray,” that is, DORV variants that have neither aorto-mitral nor pulmonary-mitral continuity and have variable distribution of conal septum (Fig. 49.1). These variations are more ambiguous both anatomically and physiologically and should be approached with an individualized management plan.
Common nomenclature for the types of DORV, for example, DORV with subaortic ventricular septal defect (VSD), DORV with subpulmonic VSD, and DORV with doubly-committed VSD, gives the impression that VSD position creates the difference, and that the relationship of the great arteries does not change (9,11). In fact, however, across the spectrum of DORV, it is the rotation of the great arteries, driven by conal development that changes, not the position of the VSD. The VSD is constant; it is almost always a typical conoventricular VSD located in the perimembranous area and extending into the trabecular ventricular septum, with the upper part of the defect bordered by the lower edge of the conus. Emphasis on the influence of infundibular development unifies the conceptual approach to the spectrum of defects that constitute DORV. Due to the degree of morphologic variability, the description of an individual patient’s heart with DORV must include the
associated relationship of the great vessels and the VSD to be most meaningful.
associated relationship of the great vessels and the VSD to be most meaningful.
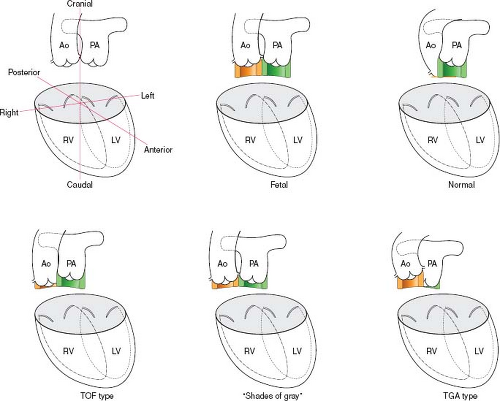 Figure 49.1 Development of the conal septum in the fetus, in the normal heart, and in different types of DORV. In the fetus, there is a circular tube of muscle, the conus, beneath each great artery. The distribution of conal muscle is equal beneath the aorta and the pulmonary artery. In the normal heart, the pulmonary valve sits up on the conus, and is positioned anteriorly and superiorly. The conal muscle beneath the aortic valve largely resorbs, leaving the aorta positioned inferiorly and posteriorly. In DORV variants, the spectrum of distribution of conal muscle beneath each great artery determines the relative position of the two semilunar valves to the ventricles. The more conal muscle present beneath a semilunar valve, the more that valve is pushed superiorly and anteriorly. “Tetralogy type”: One end of the spectrum—near-normal length of conus beneath the pulmonary valve and minimal conus beneath the aortic valve. Pulmonary valve is anterior and superior. No aorto-mitral continuity. “Shades of gray”: Between the tetralogy type and the transposition type, there are many “shades of gray”—variants with bilateral conus that have neither aorto-mitral nor pulmonary-mitral continuity and have variable distribution of conal septum. Neither great vessel is tucked in posteriorly. “Transposition type”: At the other end of the spectrum—large amount of conus under the aortic valve with relatively little under the valve. Aorta is pushed anteriorly and superiorly, resulting in rightward positioning of the aorta relative to the pulmonary artery. No pulmonary-mitral continuity. NOTE: Although two-dimensional diagrams and images demonstrate the conus as a “tear drop,” it is important to note that in three dimensions, it actually is a circular tube of muscle. Yellow, subaortic conus; green, subpulmonary conus; Ao, aorta; PA, pulmonary artery; RV, right ventricle; LV, left ventricle; TGA, transposition of the great arteries; TOF, tetralogy of Fallot. |
PS is by far the most common lesion associated with DORV. It occurs in approximately 50% of patients and may be valvar or subvalvar. Secundum atrial septal defects are seen in 25% of all types, whereas primum defects are seen in the 8% of DORV patients with AV septal defects. Multiple other associated lesions have been reported at low rates: patent ductus arteriosus, right aortic arch, subaortic stenosis, additional muscular VSDs, left superior vena cava (SVC) to the coronary sinus or left atrium, intact ventricular septum, and mitral valve anomalies including mitral atresia (9,10,20,21,22). Coronary arterial anomalies occur
in about 10% of patients—most commonly anomalous origin of the anterior descending coronary artery from the right coronary artery. However, coronary arterial anomalies are of particular importance, because they may alter considerations for surgical repair due to their effect on feasibility of conduit placement or coronary arterial transfer (23) (Fig. 49.2). Likewise, associated aortic arch coarctation, hypoplasia, or interruption—also found in about 10% of patients—significantly increase the complexity of surgical repair when present (24,25).
in about 10% of patients—most commonly anomalous origin of the anterior descending coronary artery from the right coronary artery. However, coronary arterial anomalies are of particular importance, because they may alter considerations for surgical repair due to their effect on feasibility of conduit placement or coronary arterial transfer (23) (Fig. 49.2). Likewise, associated aortic arch coarctation, hypoplasia, or interruption—also found in about 10% of patients—significantly increase the complexity of surgical repair when present (24,25).
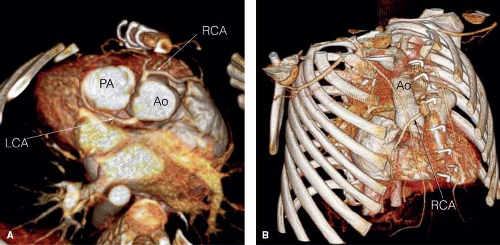 Figure 49.2 Coronary artery anomalies are of particular importance, because they may alter considerations for surgical repair due to their effect on feasibility of conduit placement or coronary transfer. CT angiograms demonstrate anomalous coronary origins and the relationship of the coronaries to the sternum well. A: CT angiogram, three-dimensional reconstruction: The right coronary artery (RCA) arises anteriorly and passes just under the sternum. LCA, left coronary artery; PA, pulmonary artery; Ao, aorta. B: CT angiogram, three-dimensional reconstruction: In a different patient, the RCA arises anomalously and crosses anterior to the aorta. Ao, aorta; RCA, right coronary artery. (Courtesy of Frandics Chan, MD, PhD, Stanford University.) |
Clinical Features: Common Variants
The clinical features and pathophysiology of DORV are determined by the morphology (22). Even within the same subtype, there can be substantial variability in the clinical presentation. Historically, cardiac catheterization with angiography and hemodynamic assessment were used routinely for diagnostic evaluation. Currently, however, transthoracic echocardiography can identify all of the essential anatomic features in most cases, and the echocardiogram along with bedside pulse oximetry provides definitive diagnosis of the pathophysiology noninvasively (26). For patients with complex aortic arch anatomy, angiography may also be needed (27,28). Magnetic resonance imaging (MRI) is helpful to further define the relationship between the VSD and the semilunar valves in cases in which it is not clear which way to baffle the VSD during reparative surgery (29,30,31,32). In a minority of patients, for example those for whom the technical feasibility of a two-ventricular repair is uncertain, cardiac catheterization is used to define pulmonary vascular resistance to determine suitability for a Fontan operation. Hemodynamic assessment via cardiac catheterization may also be necessary for another small subset of patients in whom the effects of intracardiac streaming are variable and less apparent (10).
Tetralogy Type
The most common variant is the “tetralogy of Fallot type,” with most of the conus under the pulmonary valve and minimal conal septum under the aorta (see Fig. 49.1). This variant, also known as “DORV with subaortic VSD with PS,” is seen in approximately 40% of cases (9,11). The pulmonary valve is positioned anteriorly and superiorly, and the aorta overrides the interventricular septum. Blood from the left ventricle passes through the VSD to the aorta. The cardiovascular pathophysiology is determined predominantly by the degree of PS, which generally increases over time in early infancy. Typically there is progressive, dynamic obstruction to pulmonary blood flow at the subvalvar level, leading to oxygen saturations between 80% and 90% at baseline, with further desaturation during agitation. There is the potential for hypercyanotic spells as in tetralogy of Fallot. The cardiovascular examination is notable for a long, harsh systolic ejection murmur, loudest at the left upper sternal border and caused by the subvalvar PS. Since the VSD is not pressure restrictive, there is no additional VSD murmur. Neonates with this anatomy may present with cyanosis if there is severe PS, but more often the lesion is detected due to an evaluation performed because of a murmur, or more recently, during routine neonatal oximetric examination. Cyanosis may not be present for several weeks, but after that time, it gradually worsens. The chest radiograph demonstrates normal to mildly diminished pulmonary vascular markings. The electrocardiogram is notable for right axis deviation and a right ventricular hypertrophy pattern, with rR′, qR, or rsR′ pattern; these findings are not, however, sufficiently specific to be diagnostic. Echocardiography can clearly define the essential anatomic features (Fig. 49.3). In the parasternal long-axis view, the absence of mitral-aortic continuity is shown as well as the presence of a large VSD. Subcostal views highlight
the narrow subpulmonic tunnel. The apical four-chamber view demonstrates the degree of aortic override over the crest of the interventricular septum along with the dimensions of the pathway for closure of the VSD to the aorta. Attention must be given to the chordal attachments of each AV valve and their relationships to the VSD. Computed tomography (CT), MRI, and 3-D printer casts may add to the understanding of the 3-D relationships (Figs. 49.4 and 49.5). The diagnosis may be made with fetal echocardiography; accurate assessment of the relationships of the great vessels and the VSD is possible prenatally with enough precision to guide surgical planning in most cases with tetralogy features (33,34,35).
the narrow subpulmonic tunnel. The apical four-chamber view demonstrates the degree of aortic override over the crest of the interventricular septum along with the dimensions of the pathway for closure of the VSD to the aorta. Attention must be given to the chordal attachments of each AV valve and their relationships to the VSD. Computed tomography (CT), MRI, and 3-D printer casts may add to the understanding of the 3-D relationships (Figs. 49.4 and 49.5). The diagnosis may be made with fetal echocardiography; accurate assessment of the relationships of the great vessels and the VSD is possible prenatally with enough precision to guide surgical planning in most cases with tetralogy features (33,34,35).
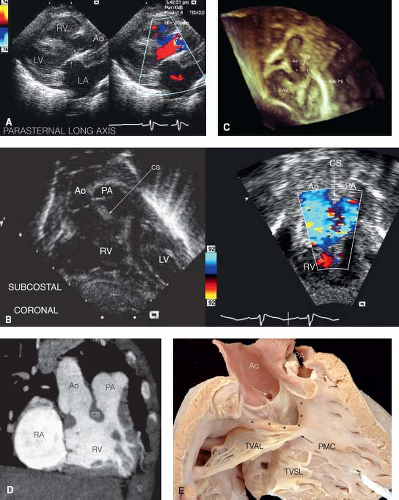 Figure 49.3 Echocardiographic views, CT angiogram image, and pathologic specimen of anatomy in “tetralogy type.” A: Echocardiogram, parasternal long-axis view: (Left panel) Note lack of mitral-aortic continuity due to conal septum beneath the aortic valve (arrows mark the conal septum) (Right panel, same image with addition of color-flow mapping). There is no flow disturbance through the ventricular septal defect, which is large and not pressure restrictive (Video 49.1). RV, right ventricle; LV, left ventricle; LA, left atrium; Ao, aorta. B: Echocardiogram, subcostal coronal view: (Left panel; Video 49.2). This image demonstrates both great arteries arising from the right ventricle, with conal septum separating the aorta and the pulmonary artery. (Right panel, same image with addition of color-flow mapping; Video 49.3.) Color-flow mapping demonstrates mild obstruction in the subvalvar area beneath the pulmonary valve due to conal muscle. RV, right ventricle; LV, left ventricle; PA, pulmonary artery; Ao, aorta; CS, conal septum. C: 3-D echocardiogram, with subaortic ventricular septal defect (asterisk) and marked subpulmonary and pulmonary valve stenosis (Video 49.4). Both great arteries arise completely from the right ventricle (RV), with aorta posterior and rightward to the pulmonary artery. The ventricular septal defect is bounded by the underside of the aortic valve and cradled inferiorly in the arms of the septomarginal trabeculations. Note the conal septum (CS) attached to the leftward, anterior division of septal band. Ao, aorta; IVS, interventricular septum; LV, left ventricle; RV, right ventricle; Sub PS, subpulmonary stenosis. D: Computed tomography angiogram, oblique image: Similar to the echo subcostal view in 3B, CT angiography demonstrates both great arteries arising from the right ventricle, with conal septum separating the aorta and the pulmonary artery. RA, right atrium; Ao, aorta; PA, pulmonary artery; CS, conal septum; RV, right ventricle. E: Pathologic specimen: The VSD is bounded by the underside of the aortic valve and cradled in the arms of the septomarginal trabeculations. The pulmonary artery is walled off by infundibular muscle. Asterisks, borders of the VSD; white arrow, coronary artery orifice; black arrow, PMC, papillary muscle of the conus; Ao, aorta; PA, pulmonary artery; TVAL, tricuspid valve anterior leaflet; TVSL, tricuspid valve septal leaflet. (C courtesy of Pierre Wong, MD, Children’s Hospital of Los Angeles; D courtesy of Frandics Chan, MD, PhD, Stanford University; E courtesy of Diane Spicer, BS, University of Florida.) |
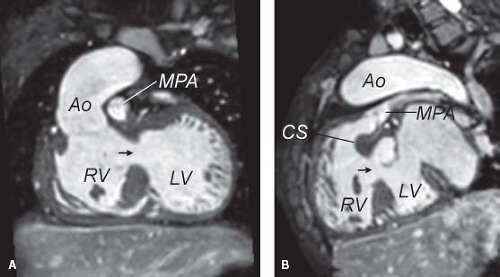 Figure 49.4 MRI images of “tetralogy type.” A: This image highlights the unobstructed pathway from the LV to the aorta, which will be surgically baffled for repair of this type of DORV. B: This sagittal image demonstrates prominent conal muscle beneath the pulmonary valve. The degree of obstruction at the subvalvar level determines the degree of cyanosis. Black arrow, Ventricular septal defect; Ao, aorta; CS, conal septum; MPA, main pulmonary artery; RV, right ventricle; LV, left ventricle. (Courtesy of Tal Geva, MD, Children’s Hospital Boston.) |
Surgery is indicated to relieve the obstruction to pulmonary flow and to septate the circulations. For this type of DORV, surgical outcomes are excellent in both neonates and older infants. Surgical repair or palliation with a modified Blalock–Thomas–Taussig shunt as a neonate is indicated when there is severe limitation to pulmonary blood flow. In most patients, however, surgery is performed electively between 2 and 4 months of age before the infant becomes excessively cyanotic or develops hypercyanotic spells.
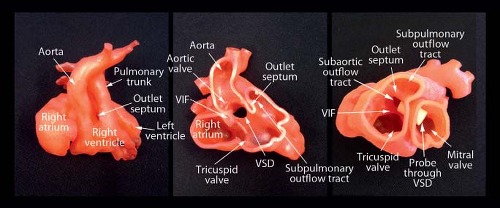 Figure 49.5 3-D printer casts of “tetralogy type.” Left panel demonstrates both great arteries arising from the right ventricle. Middle panel clearly shows the aorta overriding the interventricular septum and the VSD. In the right panel, a probe has been placed through the VSD to highlight the pathway through which blood travels from the left ventricle to the aorta. Outlet septum, conal septum; VSD, ventricular septal defect; VIF, ventriculoinfundibular fold. (Courtesy of Shi-Joon Yoo, MD, PhD, The Hospital for Sick Children, Toronto.) |
Transposition Type
The second most common variant is the “transposition type” of DORV, with a large amount of conal tissue beneath the aorta and relatively little beneath the pulmonary artery (see Fig. 49.1). Other designations for this variant, noted in approximately 20% of cases, include “Taussig–Bing anomaly” or “DORV with subpulmonic VSD” (9,11,36,37,38). In this type, the aorta is pushed anteriorly and superiorly, resulting in rightward positioning of the aorta relative to the pulmonary artery. There is usually no pulmonary-mitral continuity. The pathophysiology is determined by streaming of blood such that most of the highly oxygenated blood from the left ventricle passes through the VSD into the pulmonary artery, whereas most of the deoxygenated blood from the RV flows directly into the aorta. Thus, there is transposition physiology with a pulmonary arterial oxygen saturation that is higher than the aortic oxygen saturation.
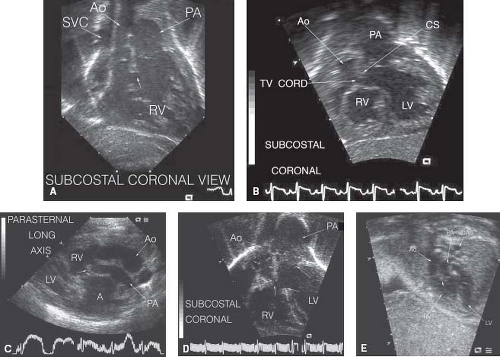 Figure 49.6 Echocardiographic views of anatomy in “transposition type.” A: Subcostal coronal view: Aorta and pulmonary artery both arise from the RV and are side by side in the same plane with the aorta rightward. Relatively large subaortic conus (small arrow) separates the aorta and pulmonary artery (Video 49.5). Aortic valve and ascending aorta are hypoplastic. SVC, superior vena cava; Ao, aorta; PA, pulmonary artery; RV, right ventricle. B: Subcostal coronal view: Aorta and pulmonary artery both arise from the right ventricle with the aorta rightward and slightly anterior (Video 49.6). Large subaortic conus separates the aorta and pulmonary artery with narrowing of the egress from the left ventricle due to the conal septum and tricuspid valve chordal attachments. RV, right ventricle; LV, left ventricle; PA, pulmonary artery; Ao, aorta; CS, conal septum; TV cord, tricuspid valve chordal attachment. C: Parasternal long-axis view: Aorta and pulmonary artery both arise from the RV with the aorta rightward and anterior (Video 49.7). There is tissue beneath the pulmonary valve (small arrow); due to this subvalvar obstruction, the pulmonary artery is relatively small compared to the aorta. RV, right ventricle; LV, left ventricle; Ao, aorta; PA, Pulmonary artery; A, atrium. D: Subcostal coronal view: Aorta and pulmonary artery both arise from the RV, side by side, in the same plane with the aorta rightward (Video 49.8). Aortic valve leaflet tips marked with small arrows. Note subaortic obstruction due to large subaortic conus (vertical arrow). Due to the subaortic obstruction, the aortic valve and ascending aorta are relatively hypoplastic. Ventricular septal defect demarcated by arrows at the crest of the interventricular septum and at the small rim of conal septum beneath the pulmonary valve. RV, right ventricle; LV, left ventricle; AO, aortic valve; PA, pulmonary artery. E: Fetal echocardiogram in Taussig–Bing heart: Aorta and pulmonary artery both arise from RV, side by side with the aorta rightward. Small arrow marks conal septum (Video 49.9). RV, right ventricle; LV, left ventricle; Ao, aorta; PA, pulmonary artery; DAO, descending aorta.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|