Chapter 58 Diseases of the Lymphatic Circulation
Anatomy of Lymphatic Circulation
Recognition of the lymphatic system and comprehension of its importance came relatively late to the medical and scientific communities. It was not until the 17th century that Gasparo Aselli recognized the lymphatics as a distinct anatomical entity.1 The gifted professor of anatomy and surgery from Milan was frequently asked by friends and colleagues to perform his elegant vivisections, and on July 23, 1622, he intended to demonstrate to them the action and innervation of the canine diaphragm.
“While I was attempting this, and for that purpose had opened the abdomen and was pulling down with my hand the intestines and stomach…I suddenly beheld a great number of cords, as it were, exceedingly thin and beautifully white, scattered over the whole of the mesentery and the intestine, and starting from almost innumerable beginnings…I noticed that the nerves belonging to the intestine were distinct from these cords, and wholly unlike them, and besides, were distributed quite separately from them. Wherefore struck by the novelty of the thing, I stood for some time silent…When I gathered my wits together for the sake of the experiment, having laid hold of a very sharp scalpel, I pricked one of these cords and indeed one of the largest of them. I had hardly touched it, when I saw a white liquid like milk or cream forthwith gush out. Seeing this, I could hardly restrain my delight.”1
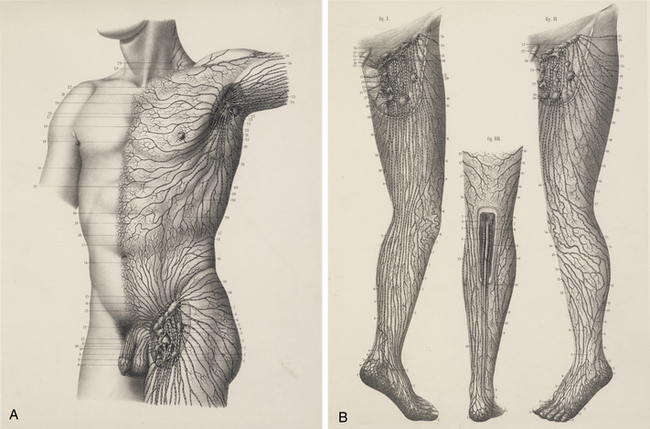
The chylous return from the intestine of the recently fed animal allowed Aselli to recognize the lymphatics; when he repeated the demonstration several days later, no vessels were to be seen. Aselli eventually realized the relation between feedings and the visibility of the mesenteric lymphatics and duplicated the work in several species (Fig. 58-1). Over the following half century, Aselli’s work was extended by Pecquet, Bartholinus, and Rudbeck, who defined the gross anatomy of the lymphatic system in toto.1 By the 18th century, smaller lymphatic channels were visualized by Anton Nuck using mercury injections. Using those techniques, Sappey observed and recorded the human lymphatic system in exquisite detail (Fig. 58-2). Even greater resolution of the anatomy was provided by von Recklinghausen in 1862 with his discovery that the lymphatic endothelium stained darkly with silver nitrate. Using that technique, he differentiated lymphatic capillaries from blood vessel capillaries. Most recently, substantial advances in the techniques of immunohistochemistry and transmission electron microscopy have enabled the certain identification of the lymphatic microcirculation and discrimination from blood vasculature.2,3
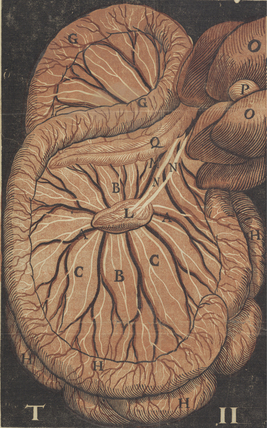
Figure 58-1 Original anatomical illustration of visceral lymphatics by Gasparo Aselli.
(From the monograph De Lactibus Sive Lacteis Venis, courtesy Harvard Medical Library, Francis A. Countway Library of Medicine.)
It is now well established that lymphatic capillaries are blind-ended tubes formed by a single layer of endothelial cells (ECs). The ECs of lymphatic capillaries closely resemble those of blood vessels and have a common embryonic origin.4 Like blood vascular endothelium, cultured lymphatic ECS form confluent “cobblestone” monolayers that “sprout” to form tubules. They demonstrate identical histological markers (von Willebrand factor (vWF), F-actin, fibronectin (FN), and Weibel-Palade bodies). Unlike systemic capillaries, the basement membrane of lymphatic capillaries is absent or widely fenestrated, allowing greater entry of interstitial proteins and particles.
The capillaries join to form larger vessels (100-200 μm) that are invested with smooth muscle and capable of vasomotion. Those vessels in turn merge to form larger conduits composed of three distinct layers: intima, media, and adventitia. Those conduits possess intraluminal valves located a few millimeters to centimeters apart that ensure lymph flow is directed centrally.5,6
Physiology of Lymphatic Circulation
In 1786, William Hunter and two of his pupils, William Cruikshank and William Hewson, published the results of their work, laying a foundation for the physiology of the lymphatic system.1 They correctly inferred from clinical observation that the lymphatics were involved in the response to infection, as well as in the absorption of interstitial fluid. A century later, their theories received experimental support from the physiological studies of Karl Ludwig and Ernest Starling. Ludwig cannulated lymph vessels, collected and analyzed the lymph, and proposed that it was a filtrate of plasma. Starling elucidated the forces governing fluid transfer from the blood capillaries to the interstitial space and offered evidence that the same forces apply to the lymphatic capillaries. He proposed that an imbalance in those forces could give rise to edema formation:
“In health, therefore, the two processes, lymph production and absorption are exactly proportional. Dropsy depends on a loss of balance between these two processes—on an excess of lymph-production over lymph-absorption. A scientific investigation of the causation of dropsy will therefore involve, in the first place, an examination of the factors which determine the extent of these two processes and, so far as is possible, the manner in which these processes are carried out.”7
As first enunciated by Starling, interstitial fluid is largely an ultrafiltrate of blood. Its rate of production reflects the balance between factors that favor filtration out of capillaries (capillary hydrostatic pressure and tissue oncotic pressure) and those that favor reabsorption (interstitial hydrostatic pressure and capillary oncotic pressure).7 Under normal conditions, filtration exceeds reabsorption at a rate sufficient to create 2 to 4 L of interstitial fluid per day. There is a net filtration of protein (primarily albumin) from the vasculature into the interstitium; approximately 100 g of circulating protein may escape into the interstitial space daily. The interstitial fluid also receives the waste products of cellular metabolism, as well as foreign matter or microbes that enter through breaks in the skin or by hematogenous routes.
Not surprisingly, the lymphatics require a complex interplay of specific anatomy and function to meet physiological requirements. Several forces drive fluid through the lymphatic system. The lymphatic vessels in skeletal muscle are compressed by extrinsic muscular contractions that propel the fluid centrally through the unidirectional valves. In other tissues, such as the splanchnic and cutaneous systems, it is primarily contractions of lymphatic smooth muscle that generate the driving force.8,9 These contractions are increased in frequency and amplitude by elevated filling pressure, sympathetic nerve activity, and shock, and they may be modulated by circulating hormones and prostanoids.10–12 Considerable force can be generated by those contractions; experimentally induced obstruction of the popliteal lymphatic system augments the strength and frequency of contraction, generating pressures of up to 50 mmHg.13 Other factors that may contribute to lymphatic flow include intermittent compression from arterial pulsations and gastrointestinal peristalsis. In addition, it has most recently been proposed that the initial lymphatics (small lymphatic capillaries that begin blindly in the tissues) most likely possess a two-valve system.14,15 In addition to the classically described secondary intralymphatic valves, the initial lymphatics are thought to possess a primary valve system at the level of the endothelium to ensure unidirectional flow at this level. Once lymph enters the thorax, negative intrathoracic pressure generated during inspiration aspirates fluid into the thoracic duct (the “respiratory pump”).13 Failure of adequate lymph transport promotes lymphedema and likely contributes to the pathological presentation of a wide variety of lymphatic vascular diseases.
Lymphatic Insufficiency (Lymphedema)
Pathogenesis of Edema
Edema develops when production of interstitial fluid (lymph) exceeds its transport through the lymphatic system. Thus, overproduction of lymph (enhanced lymphatic load) or decreased ability to remove fluid (defective transport) from the interstitium may result in edema. Conditions associated with overproduction of lymph include elevated venous pressures, increased capillary permeability, and hypoproteinemia. Elevated hydrostatic pressure in the veins results in increased filtration of plasma from venules and blood capillaries (as seen in right-sided congestive heart failure (CHF), tricuspid regurgitation, and deep vein thrombosis [DVT]). Conversely, local inflammation increases capillary permeability, accelerating loss of protein and fluid to the interstitium despite a normal capillary hydrostatic pressure. Lymph production may increase by 10- to 20-fold, exceeding lymphatic transport and resulting in marked edema.16 Hypoproteinemia also may lead to marked edema, in which case hydrostatic pressure and capillary permeability are normal, but capillary oncotic pressure is reduced, favoring osmotic flow to the interstitium. The edema that ensues in these conditions can, strictly speaking, be called lymphedema only when there is objective evidence of impaired lymphatic clearance or physical evidence of consequences of impaired lymphatic function in the skin or subcutaneous tissues.
Pathogenesis of Lymphedema
Primary lymphedema
Prevalence estimates for the heritable causes of lymphedema are difficult to ascertain and vary substantially. Primary lymphedema is thought to occur in approximately 1 of every 6 to 10,000 live births. Females are affected 2- to 10-fold more commonly than males.17,18 Primary lymphedema represents a heterogeneous group of disorders, and therefore its classification schemata are numerous. Affected individuals can be classified by age of onset, functional anatomical attributes, or clinical setting.
Age of Onset
When distinguished by age of clinical onset, primary lymphedema can typically be divided into the following categories19:
Anatomical Patterns
An alternative classification scheme relies on an anatomical description of the lymphatic vasculature20,21:
1. Aplasia: no collecting vessels identified.
2. Hypoplasia: a diminished number of vessels are seen.
3. Numerical hyperplasia (as defined by Kinmonth): an increased number of vessels are seen.
4. Hyperplasia: in addition to an increase in number, the vessels have valvular incompetence and display tortuosity and dilation (megalymphatics).
Approximately one third of all cases are secondary to agenesis, hypoplasia, or obstruction of the distal lymphatic vessels, with relatively normal proximal vessels.26,27 In those cases, swelling is usually bilateral and mild and affects females much more frequently than males. The prognosis in such cases is good. Generally after the first year of symptoms, there is little extension in the same limb or to uninvolved extremities. Although the extent of involvement is established early in the disease in about 40% of patients, the girth of the limb continues to increase.
In more than half of all cases, the defect primarily involves obstruction of the proximal lymphatics or nodes, with initial lack of involvement of distal lymphatic vessels. Pathological studies reveal intranodal fibrosis.15 In those cases, swelling tends to be unilateral and severe; there may be a slight predominance of females in this group. In patients with proximal involvement, the extent and degree of the abnormality is more likely to progress and require surgical intervention. Initially uninvolved distal lymphatic vessels may become obliterated with time.
Clinical Characteristics
As a third alternative, the primary lymphedemas can often be characterized by associated clinical anomalies or abnormal phenotype.21 Although sporadic instances of primary lymphedema are more common, the tendency for congenital lymphedema to cluster in families is significant. The syndrome of a familial predisposition to congenital lymphedema, ultimately determined to ensue from an autosomal dominant form of inheritance with variable penetrance, was first described by Milroy in 1892.22 He reported “hereditary edema” affecting 22 individuals of one family over six generations. Although Milroy ultimately came to consider not only congenital lymphedema but also praecox and tarda forms as variants of the syndrome that bears his name,23 the praecox form of primary lymphedema more often carries the eponym of Meige’s disease.24
In fact, a long list of disorders are associated with heritable forms of lymphedema. Increasingly these disorders have yielded to chromosomal mapping techniques. Lymphedema-cholestasis, or Aagenaes’ syndrome, has been mapped to chromosome 15q.25 In several family cohorts of Milroy’s disease, it has been determined that the disorder reflects missense inactivating mutations in the tyrosine kinase domain of vascular endothelial growth factor receptor 3 (VEGFR3),26,27 thus underscoring the likelihood that the pathogenesis of this condition likely reflects an inherited defect in lymphatic vasculogenesis. Several additional lymphedema syndromes have recently lent themselves to successful genetic mapping.21 Lymphedema-distichiasis, an autosomal dominant dysmorphic syndrome in which lymphedema presents in association with a supplementary row of eyelashes arising from the meibomian glands, has been linked to truncating mutations in the forkhead-related transcription factor FOXC228; mutations in FOXC2 have subsequently been associated with a broad variety of primary lymphedema presentations.29 Similarly, a more unusual form of congenital lymphedema, hypotrichosis-lymphedema-telangiectasia, has been ascribed to both recessive and dominant forms of inheritance of mutations in the transcription factor gene SOX18.30 Most recently, linkage analysis of three affected family cohorts has associated the occurrence of autosomal recessive congenital lymphatic dysplasia (Hennekam’s syndrome) to the gene CCBE1,31,32 also identified as critical to lymphangiogenesis in zebrafish.31 It is altogether plausible that further elucidation of the molecular pathogenesis of these diseases linked to FOXC2, SOX18, and CCBE1 mutations will lead to enhanced insights into mechanisms of normal and abnormal lymphatic development. Furthermore, mutational analysis of families expressing inherited forms of lymphedema have disclosed specific mutations in hepatocyte growth factor (HGF) (which encodes HGF) and MET (the HGF receptor).33 GJC2, the gene that encodes connexin-47 has also been implicated in the familial occurrence of lymphedema.34
In general, autosomal or sex-linked recessive forms of congenital lymphedema occur less commonly than the dominant forms of inheritance. Nevertheless, the list of heritable lymphedema-associated syndromes is long and growing (Box 58-1). Primary lymphedema has been described in association with various forms of chromosomal aneuploidy (e.g., Turner’s and Klinefelter’s syndromes), various dysmorphogenic genetic anomalies (e.g., Noonan’s syndrome, neurofibromatosis), and with various as yet unrelated disorders such as yellow nail syndrome, intestinal lymphangiectasia, lymphangiomyomatosis, and arteriovenous malformation (AVM).35 The association of lymphedema with vascular anomalies likely derives from the shared embryological origin of the lymphatic and venous vasculature.
 Box 58-1 Hereditary Conditions Associated with Lymphedema
Box 58-1 Hereditary Conditions Associated with Lymphedema
Dysmorphogenic-Genetic Disturbances
From Rockson SG: Syndromic lymphedema: keys to the kingdom of lymphatic structure and function? Lymph Res Biol 1:181, 2003.21
Secondary lymphedema
Infection
Recurrent bacterial lymphangitis leads to thrombosis and fibrosis of the lymphatic channels and is one of the most common causes of lymphedema.36 The responsible bacteria are almost always streptococci, which tend to enter through breaks in the skin or fissures induced by trichophytosis. Recurrent bacterial lymphangitis is also a frequent complicating factor of lymphedema from any cause.
The microfilaria are transmitted by a mosquito vector and induce recurrent lymphangitis and eventual fibrosis of lymph nodes. It is unclear whether filaria themselves produce the lymphangitis or simply predispose those afflicted to recurrent episodes of bacterial lymphangitis. The filaria also can be identified in blood specimens of tissue obtained by fine-needle biopsy of affected areas, and eosinophilia is a common local and systemic feature. Diethylcarbamazine remains the most popular drug for treating filariasis; although side effects are frequent, it is extremely efficacious.37 Ivermectin is a newer antifilarial agent that may replace diethylcarbamazine; it is less toxic, and a single oral dose (25 μg/kg) appears to be as efficacious as a 2-week course of diethylcarbamazine.38
Lymphatic Trauma
Within this category, by far the most common mechanism of lymphedema relates to surgical excision of lymph nodes.39 This occurs most commonly in the setting of cancer staging and therapeutics. Breast cancer–associated lymphedema is the most common form of lymphedema in the United States. Both axillary lymph node dissection and adjuvant radiation therapy, particularly to the breast and axilla, predispose to development of secondary lymphedema of the upper extremity.40 In clinical follow-up after periods of up to 13 years following invasive treatment of breast cancer, a late incidence of lymphedema of 14% can be expected in surgically treated patients with adjuvant postoperative irradiation.41 Even less radical surgery (e.g., lumpectomy) can occasionally be complicated by lymphedema. Despite improvements in surgical and radiotherapeutic techniques, lymphedema remains a potential complication.42,43 Similarly, edema of the leg may occur after surgery for pelvic or genital cancer, particularly when there has been inguinal and pelvic lymph node dissection or irradiation.44 Its reported frequency varies between 1% and 47%.45,46 Pelvic irradiation increases the frequency of leg lymphedema after cancer surgery47 for such conditions as malignant melanoma, prostate cancer, and gynecological malignancies.
Lymphedema can also ensue from other mechanisms of lymphatic trauma, among them burns, large or circumferential wounds of the extremity, or other iatrogenic causes.48
Other Causes
Other conditions leading to or associated with obstruction of lymphatic channels include tuberculosis, contact dermatitis, rheumatoid arthritis, and pregnancy.49 Autoimmune destruction of the lymphatics remains an interesting but unproven etiology.50 Factitious lymphedema (“oedema bleu”) also occurs. The condition usually affects the hand, arm, or both; is unilateral; and is induced by applications of tourniquets, self-inflicted cellulitis, or maintenance of the limb in an immobile and dependent state. Chronic subcutaneous injections of drugs (most notably, pentazocine hydrochloride) also may lead to lymphatic sclerosis and obstruction.
Pathology of Lymphedema
Early in the natural history of lymphedema, there is variable thickening of the epidermis; the skin becomes rough with hyperkeratosis and accentuation of skin folds. Organization and fibrosis may lead to development of papillomatosis. Early on, substantial edema of the dermis may occur. On cut section of gross specimens, the dermis is firm and gray, as is the deep fascia. Usually later in the course, but sometimes quite early, there may be expansion of the subcutaneous adipose tissue, often septated by prominent fibrous strands. In some specimens, compression causes lymph to exude from the dermis and subcutaneous tissue, although this is not a prominent feature. Microscopic examination reveals hyperkeratosis with prominent dermal fibrosis, as well as variable degrees of dermal edema (Fig. 58-3). Abundant subcutaneous fat with prominent fibrous septa is apparent in all cases. Often, perivascular inflammatory cells (lymphocytes, plasma cells, and occasionally eosinophils) can be seen. The lymphatic vessels often are difficult to visualize and may be obliterated or thrombosed by previous inflammatory episodes or may be congenitally absent or hypoplastic. Dilation of the lymphatics may be seen.
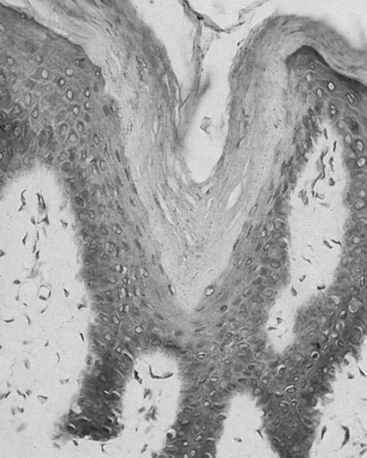
Figure 58-3 Skin biopsy in chronic lymphedema discloses characteristic hyperkeratosis and hypercellularity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


