Dilated Cardiomyopathy
Kimberly Y. Lin
Joseph W. Rossano
Introduction
Dilated cardiomyopathy (DCM) is a heart muscle disorder characterized by left ventricular (LV) dilation and systolic dysfunction, often resulting in low cardiac output secondary to heart failure. It is the most common cardiomyopathy in children, with an estimated incidence of 0.57 cases per 100,000 children per year (1). While some children who present with DCM show normalization of their heart size and function over time, the disease carries a high risk of morbidity and mortality with many patients needing advanced heart failure therapies including mechanical circulatory support and heart transplantation.
Etiologies
There are multiple underlying diseases that can lead to the phenotype of a dilated left ventricle with depressed ventricular function (Table 53.1). The concept of a “final common pathway” was proposed by Towbin and Belmont in the late 1990s and hypothesized that cytoskeletal abnormalities and disruption in the linkage of the sarcomere to the sarcolemma would be the issue underlying the pathogenesis of various disease processes leading to DCM (2,3). This notion of a final common pathway for diverse etiologies leading to a common phenotype has been expanded to include other diseases including hypertrophic cardiomyopathy, restrictive cardiomyopathy, arrhythmogenic ventricular cardiomyopathy, LV noncompaction, and arrhythmogenic disorders (Fig. 53.1) (4,5,6,7). This
hypothesis led to targeted investigation and discovery of multiple genes that can lead to similar phenotypes and sometimes a phenotype that overlaps with other cardiomyopathies. Genetic abnormalities that have been associated with DCM are listed in Table 53.2.
hypothesis led to targeted investigation and discovery of multiple genes that can lead to similar phenotypes and sometimes a phenotype that overlaps with other cardiomyopathies. Genetic abnormalities that have been associated with DCM are listed in Table 53.2.
TABLE 53.1 Etiologies of Dilated Cardiomyopathy | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
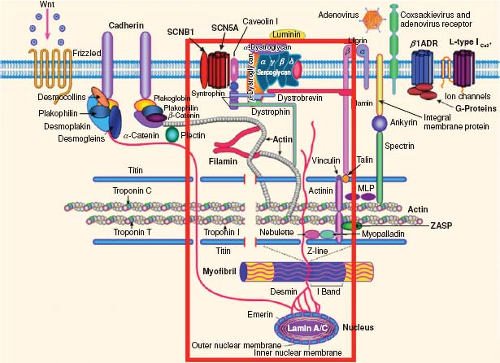 Figure 53.1 Cardiomyocyte demonstrating abnormalities that can lead to cardiomyopathy. In this illustration of the cardiomyocyte, the extracellular matrix (in which laminin is shown), sarcolemma (in which SCN5A is shown), sarcomere (in which the troponins are shown), and nucleus (in which lamin A/C is shown) are shown along with a variety of interactive proteins within each area of the cardiomyocyte. The cardiac sodium channel, SCN5A, is shown in the sarcolemma and interacts with its subunit SCNB1 in the sarcolemma, as well as dystrophin, syntrophin, and caveolin, among other proteins (note red rectangular box denoting this pathway). As can be seen, disturbance of any of these interacting proteins can disrupt the function of the others that interact directly or downstream, thereby resulting in dysfunction and a clinical cardiac phenotype such as dilated cardiomyopathy or arrhythmias, or both. (From Towbin JA, Lorts A. Arrhythmias and dilated cardiomyopathy common pathogenetic pathways? J Am Coll Cardiol. 2011;57:2169–2171.) |
TABLE 53.2 Genetics of Dilated Cardiomyopathy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Pathophysiology
In the neurohormonal model of heart failure, there is an index injury event to the myocardium leading to loss of myocytes and LV dysfunction (8). This leads to activation of the renin–angiotensin–aldosterone system, the adrenergic nervous system, and a variety of inflammatory cytokines. As heart failure is a systemic syndrome, multiple other biochemical abnormalities including vascular dysfunction, renal dysfunction, and oxidative stress also occur (9). Many of these responses are initially compensatory, though are maladaptive in the chronic state and lead to more myocardial stress, LV dilation, and eventually end-stage heart failure (Fig. 53.2) (8). Indeed, blocking the maladaptive response of the renin–angiotensin–aldosterone system and adrenergic nervous system pharmacologically has led to substantially improved survival in adults with heart failure (10).
While in adults the most common causes of DCM are ischemia and hypertension, in children the majority of cases are idiopathic. However, an investigation into the potential causes of LV dilation and dysfunction is warranted, as some of the underlying etiologies are potentially treatable, or may have multisystem organ involvement. Sarcomere or cytoskeletal gene mutations are currently identifiable in approximately 20% to 35% of children and adults with DCM (11,12,13). Other potential causes of DCM in children include inflammatory disorders such as myocarditis, toxic exposures such as anthracycline therapy, and ischemia, volume overload, or pressure overload from congenital heart lesions. DCM may also be one component of a systemic disorder including various metabolic, mitochondrial, and neuromuscular diseases.
Histologically, patients with advanced heart failure from DCM often have varying degrees of myofibrillar loss with replacement
fibrosis, myocyte hypertrophy, and disruption of the normal cytoskeletal architecture (14,15,16,17,18). Abnormal mitochondria are often present, even in the absence of primary mitochondrial disorders (14,17,18). Some of these findings are improved, at least temporarily, with unloading the ventricle with a ventricular assist device (15,16). Additionally, evidence of inflammation may be identified in those with underlying myocarditis.
fibrosis, myocyte hypertrophy, and disruption of the normal cytoskeletal architecture (14,15,16,17,18). Abnormal mitochondria are often present, even in the absence of primary mitochondrial disorders (14,17,18). Some of these findings are improved, at least temporarily, with unloading the ventricle with a ventricular assist device (15,16). Additionally, evidence of inflammation may be identified in those with underlying myocarditis.
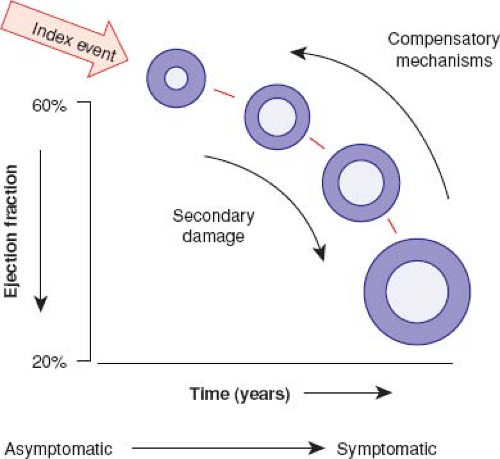 Figure 53.2 Pathogenesis of heart failure. Heart failure begins after an index event produces an initial decline in pumping capacity of the heart. After this initial decline in pumping capacity of the heart, a variety of compensatory mechanisms are activated, including the adrenergic nervous system, the renin–angiotensin system, and the cytokine system. In the short term these systems are able to restore cardiovascular function to a normal homeostatic range, with the result that the patient remains asymptomatic. However, with time the sustained activation of these systems can lead to secondary end-organ damage within the ventricle, with worsening LV remodeling and subsequent cardiac decompensation. As a result of resultant worsening LV remodeling and cardiac decompensation, patients undergo the transition from asymptomatic to symptomatic heart failure. (From Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:1837–2894.) |
At a cellular level, myocyte injury, hypertrophy, death, and fibrosis lead to a loss of myocardial contractile function. Regardless of the etiology of DCM, impaired myocardial function stimulates neurohormonal compensatory mechanisms in an attempt to maintain cardiac output in the setting of decreased contractile function. This response, while initially compensatory, perpetuates a pathologic cascade of cardiac remodeling that results in ventricular dilation, fibrosis, and further dysfunction. Associated atrial or ventricular arrhythmias, mitral and/or tricuspid regurgitation, intracavitary thrombi, and hypoperfusion can lead to end-organ damage.
Clinical Evaluation
There is a wide range of initial presentations for a child with DCM. Some patients are completely asymptomatic, as is often the case in those undergoing screening in the setting of a neuromuscular disorder or a known family history of cardiomyopathy. Others may present in florid heart failure, perhaps triggered by a stressor such as a febrile illness, exercise, or respiratory infection.
When presented with a new diagnosis of DCM, any potentially correctable structural cardiac defects must be ruled out before labeling the child with a primary heart muscle disorder. Congenital coronary anomalies, such as anomalous origin of the left coronary artery from the pulmonary (ALCAPA) that commonly presents with severe heart failure in infancy, must be part of the diagnostic evaluation as there is a favorable long-term outcome with surgical correction. One should also try to establish the likelihood of an acute inflammatory process (such as myocarditis) and presence of any related systemic disorders, as these may affect the treatment options and prognosis.
History and Physical Examination
Patient’s histories with DCM are quite variable. Infants and young children may have poor growth. Multiorgan system involvement is not rare, especially for mitochondrial or metabolic disorders. Patients with neuromuscular disorders, such as Duchenne muscular dystrophy, demonstrate features characteristic of their systemic myopathy. Those identified while still asymptomatic are often diagnosed in the context of an abnormal ECG, chest x-ray, or screening for a family history of cardiomyopathy. However, many patients with DCM will present with symptomatic heart failure requiring resuscitation and inpatient management (1,19,20).
Congestion and/or poor perfusion are responsible for symptoms in patients with acute heart failure. Presenting symptoms can range from mild respiratory or gastrointestinal symptoms to profound cardiac shock (19,21). Other common symptoms include fatigue, nausea, vomiting, abdominal pain, chest pain, and diaphoresis. At least one gastrointestinal symptom was evident in over 80% of children in one study (21). Severe symptoms of poor perfusion and cardiogenic shock are also not uncommon (19).
The physical examination is also highly variable. Among patients in a compensated state of heart failure, the physical examination may be remarkably benign with normal heart sounds, normal pulmonary examination, and no peripheral edema or organomegaly. Among patients with symptomatic heart failure, abnormal heart sounds such as a gallop rhythm, a murmur of mitral valve regurgitation, tachypnea with rales, and tachycardia are common features. For severely symptomatic patients, poor perfusion may be evident. Unlike adults, infants and young children rarely have peripheral edema. As DCM may occur as part of a systemic disease, such as hypothyroidism, stigmata of the underlying disease should be assessed.
Diagnostic Testing
Imaging studies that are indicated for the diagnosis and initial management of patients with DCM include chest radiography, ECG, and echocardiogram. Cardiac magnetic resonance (CMR) imaging is becoming more frequently utilized and may have additional diagnostic and prognostic value. Chest x-rays generally demonstrate cardiomegaly with varying degrees of pulmonary edema. The left mainstem bronchus may be compressed by an enlarged left atrium, resulting in left lower lobe atelectasis (22). ECGs are usually abnormal in patients with DCM, especially among patients with severe
disease (Fig. 53.3). Characteristic ECG abnormalities include LV hypertrophy and ST-segment and T-wave changes (23,24). These ECG findings are not specific for DCM; however, certain abnormalities such as conduction disease in DCM are associated with LMNA gene mutations (25). Patient with myocarditis may present with ECG abnormalities such as tachyarrhythmias, conduction disease, low voltages, ST-segment changes, T-wave abnormalities, and pathologic Q waves (26,27,28).
disease (Fig. 53.3). Characteristic ECG abnormalities include LV hypertrophy and ST-segment and T-wave changes (23,24). These ECG findings are not specific for DCM; however, certain abnormalities such as conduction disease in DCM are associated with LMNA gene mutations (25). Patient with myocarditis may present with ECG abnormalities such as tachyarrhythmias, conduction disease, low voltages, ST-segment changes, T-wave abnormalities, and pathologic Q waves (26,27,28).
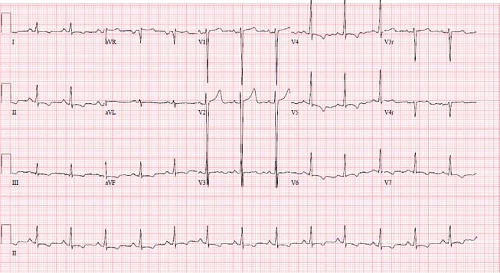 Figure 53.3 Typical electrocardiogram in a child with dilated cardiomyopathy. Note the T-wave inversions in multiple leads, and the deep S wave in V1 consistent with a left ventricular hypertrophy pattern. |
At most institutions, echocardiography is still the imaging modality of choice to establish the diagnosis of DCM, with commonly accepted diagnostic criteria in pediatric patients being a LV end-diastolic dimension >2 standard deviations above normal for body surface area, in conjunction with depressed systolic function (e.g., shortening fraction/ ejection fraction <2 standard deviations below normal for age) (Fig. 53.4 and Video 53.1) (29). In addition to being able to quantitate the degree of LV dilation
and systolic dysfunction, other important echocardiographic findings include the degree of mitral valve regurgitation, diastolic dysfunction, right ventricular involvement, and pulmonary artery pressure estimates. These may have prognostic value in patients with DCM (30,31,32,33,34).
and systolic dysfunction, other important echocardiographic findings include the degree of mitral valve regurgitation, diastolic dysfunction, right ventricular involvement, and pulmonary artery pressure estimates. These may have prognostic value in patients with DCM (30,31,32,33,34).
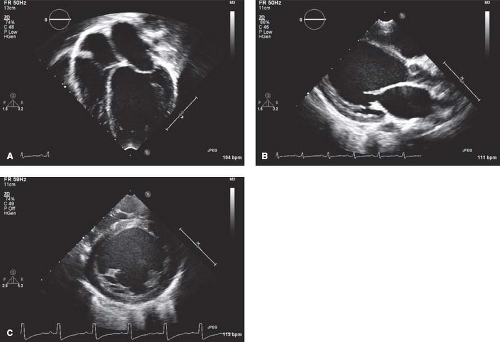 Figure 53.4 Typical echocardiographic images from a child with dilated cardiomyopathy, showing a left ventricular end-diastolic dimension >2 standard deviations above the norm for body surface area in (A) apical four-chamber, (B) parasternal long-axis, and (C) parasternal short-axis views. |
CMR is increasingly utilized in the diagnosis and management of patients with DCM, providing an alternate means of assessing ventricular volumes and function (Fig. 53.5A–C and Video 53.2) (31,35,36). The CMR can also assist in identification of underlying disease processes leading to DCM. The Lake Louise criteria are
often utilized for the diagnosis of myocarditis (Table 53.3) (36). However, while CMR can be helpful in distinguishing myocarditis from other etiologies of DCM (37,38), myocardial biopsy is still considered the gold standard and can be useful for patients with unexplained heart failure (36,39). Additional details that can be obtained by CMR include evidence of focal fibrosis by late gadolinium enhancement (Fig. 53.5D–G) or diffuse fibrosis by T1 mapping (35,40,41,42). These findings may have important prognostic implications, though further study is needed. CMR can also be useful in identification of infiltrative disorders, such as hemochromatosis, sarcoidosis, and Wegener granulomatosis (36,43).
often utilized for the diagnosis of myocarditis (Table 53.3) (36). However, while CMR can be helpful in distinguishing myocarditis from other etiologies of DCM (37,38), myocardial biopsy is still considered the gold standard and can be useful for patients with unexplained heart failure (36,39). Additional details that can be obtained by CMR include evidence of focal fibrosis by late gadolinium enhancement (Fig. 53.5D–G) or diffuse fibrosis by T1 mapping (35,40,41,42). These findings may have important prognostic implications, though further study is needed. CMR can also be useful in identification of infiltrative disorders, such as hemochromatosis, sarcoidosis, and Wegener granulomatosis (36,43).
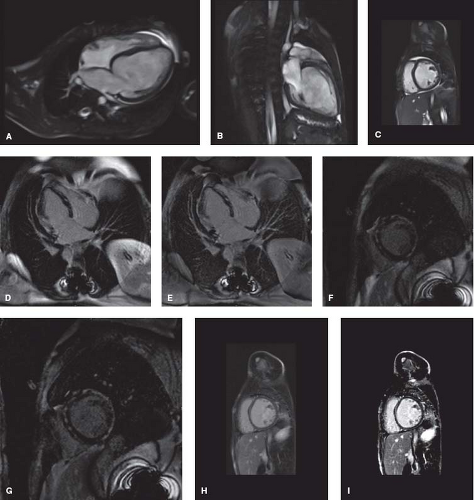 Figure 53.5 Cardiac magnetic resonance imaging in children with dilated cardiomyopathy. Steady-state free precession cine imaging demonstrates left ventricular dilation in the (A) four-chamber, (B) long-axis, and (C) short-axis views. Evidence of patchy focal fibrosis via late gadolinium enhancement is seen throughout the myocardium in a child with Duchenne muscular dystrophy on (D) magnitude inversion recovery and (E) phase-sensitive imaging in a four-chamber view, and (F) magnitude inversion recovery and (G) phase-sensitive imaging in a short-axis view. A midmuscular stripe of late gadolinium enhancement in the interventricular septum is seen in a child with idiopathic dilated cardiomyopathy via (H) magnitude inversion recovery and (I) phase-sensitive imaging in a short-axis view. |
TABLE 53.3 Proposed Cardiac Magnetic Resonance (CMR) Imaging Criteria for the Diagnosis of Myocarditis | ||
|---|---|---|
|
Laboratory assessments are important in establishing the underlying diagnosis, assessing the severity of heart failure and multiorgan dysfunction, and monitoring response to therapies. For patients with symptoms consistent with heart failure, initial laboratory testing with natriuretic peptides can help to identify patients with heart failure. B-type natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT-BNP) levels are often elevated compared to healthy controls among pediatric patients with acute noncardiac conditions such as sepsis, dehydration, and respiratory disease (44,45,46,47,48,49); however, the degree of elevation is typically greater among patients with heart failure (44,45,46,49). The prognostic value of natriuretic peptides at the time of hospital admission is unclear. In adults, higher admission levels are associated with worse outcomes; however, admission BNP or NT-BNP levels may not correlate with hospital outcomes in children (50). Some studies have found the degree of BNP elevation predictive of adverse events (51), while others have not (50,52). Serial levels may be of more value, and one study has found an association of serial NT-BNP levels and the need for mechanical circulatory support (50). An additional study found that among heart failure patients admitted to an ICU, the discharge BNP level had more prognostic value (52). Additionally, more than 80% of patients that experienced an increase in the BNP level with treatment died or were readmitted to the hospital within 60 days (52). Other testing on initial presentation of acute heart failure can vary depending on the severity of decompensation. Among adults with acute heart failure, adverse outcomes are associated with worsening renal function, hyponatremia, anemia, and increased bilirubin levels (53). It is unknown if these markers have the same prognostic value in children, although worsening renal function has certainly been associated with poor short-term outcomes in children hospitalized with heart failure (54).
A thorough assessment for underlying etiologies is of critical value in these patients. As the etiologies for DCM are diverse (see Table 53.1), testing can be tailored for the individual patient and presenting symptomatology. For example, a male patient with neutropenia should be assessed for Barth syndrome (55). Evaluating for the presence of metabolic and mitochondrial abnormalities should be part of a standard evaluation in a child with DCM. Genetic testing is also frequently performed for patients without a clear etiology and is recommended in specific contexts by many organizations including the International Society for Heart and Lung Transplantation and the Heart Failure Society of America (56,57). While the overall reported diagnostic yield for DCM is low, centers that perform comprehensive testing including the use of commercially available genetic tests have reported diagnostic yields for DCM of over 50% (13).
For patients with DCM and new-onset acute heart failure, cardiac catheterization and endomyocardial biopsy should be carefully considered (39,56,58). Hemodynamics can often be useful in patient management and invasive hemodynamic measurements including pulmonary artery pressures and pulmonary vascular resistance would be considered standard in DCM patients that are being considered for heart transplantation (59). The endomyocardial biopsy can help in establishing the underlying disorder and may be particularly helpful in challenging cases. In one study of adults with a diagnosis of new-onset heart failure undergoing endomyocardial biopsy, the biopsy revealed the underlying disease in 25% of the cases (35% if symptoms were <2 weeks of duration) and clinical management was changed in 23% of the patients based on the results (39). The risk of any type of invasive procedure must obviously be weighed against the potential benefit of the study, but in experienced centers, endomyocardial biopsies can be performed safely even in small, critically ill children (60,61,62).
An important part of testing patients with DCM is family screening. The Heart Failure Society of America recommends a clinical screening consisting of history, physical examination, electrocardiogram, echocardiogram, and creatine kinase (CK–MM isoform) measurement for first-degree family members of patients with DCM (57). If there is no evidence of disease in the family members, it is recommended that clinical screening be continued at intermittent intervals, the frequency of which depends on whether an underlying disease-causing mutation is found (Table 53.4).
Treatment
Disease-Specific Therapies
For many patients with DCM the etiology is either idiopathic or when known, there is often no disease-specific therapy available. When a child with DCM presents in heart failure, the immediate goals of care must be to manage symptoms. However, one should simultaneously
look for secondary causes of DCM where disease-specific therapies may be possible. For example, a child with previously undiagnosed ALCAPA warrants surgical repair of the coronary anomaly, and some of the rare inborn errors of metabolism may correct with carnitine supplementation. Arrhythmias are often associated with DCM, but can occasionally be the underlying cause of a cardiomyopathy, in which case treatment of the underlying arrhythmia is paramount to cardiac recovery. Interestingly, there is considerable debate surrounding the utility of immunomodulatory agents in cases where myocarditis is suspected (63,64,65,66,67,68,69,70,71). Even though there are limited data to support the use of intravenous immunoglobulin (IVIG), it is utilized in the majority of pediatric patients with acute myocarditis in the United States (72). However, the recent pediatric heart failure guidelines from the International Society for Heart and Lung Transplantation do not recommend routine administration of corticosteroids or IVIG for myocarditis (56).
look for secondary causes of DCM where disease-specific therapies may be possible. For example, a child with previously undiagnosed ALCAPA warrants surgical repair of the coronary anomaly, and some of the rare inborn errors of metabolism may correct with carnitine supplementation. Arrhythmias are often associated with DCM, but can occasionally be the underlying cause of a cardiomyopathy, in which case treatment of the underlying arrhythmia is paramount to cardiac recovery. Interestingly, there is considerable debate surrounding the utility of immunomodulatory agents in cases where myocarditis is suspected (63,64,65,66,67,68,69,70,71). Even though there are limited data to support the use of intravenous immunoglobulin (IVIG), it is utilized in the majority of pediatric patients with acute myocarditis in the United States (72). However, the recent pediatric heart failure guidelines from the International Society for Heart and Lung Transplantation do not recommend routine administration of corticosteroids or IVIG for myocarditis (56).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


