Examples of known causes
Disorders of mucociliary clearance
Cystic fibrosis
Primary ciliary dyskinesia
Young’s syndrome
Alpha-1 antitrypsin deficiency
Primary immunodeficiency
Hypogammaglobulinaemia
Common variable immunodeficiency
X-linked agammaglobulinemia
IgA deficiency
IgG subclass deficiency
Neutrophil deficiency
Chronic granulomatous disease
Schwachman-Bodian-Diamond syndrome
Innate immunity
Complement deficiency
Collagen disorders
Marfan syndrome
Williams-Campbell syndrome
Mounier-Kuhn syndrome
Other associations
Autoimmune disease
Disorders of Mucociliary Clearance
Ciliated respiratory epithelium lines the airways. The cilia, bathed in perciliary fluid, beat in a coordinated fashion at 11–18 Hz, to propel the overlying mucus along with particles and bacteria to the oropharynx where it can be swallowed or expectorated (Fig. 4.1). Diseases affecting ciliary function, or that change the composition of the periciliary fluid and mucus can impair mucociliary clearance, leading to recurrent infections and inflammation which predispose to bronchiectasis.
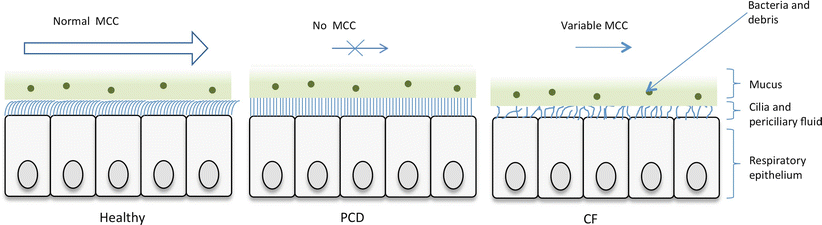
Fig. 4.1
In healthy persons, respiratory cilia beat in a coordinated sweeping pattern, which moves mucus and debris, including pathogens towards the oropharynx for swallowing or expectorating. In PCD, immotile or dyskinetic cilia do not beat effectively, and mucus and debris persist in the airways. In CF the inefficient mucociliary clearance is due to an abnormal periciliary fluid layer compromising ciliary beating, and viscous mucus which is resistant to clearance (Image provided by Robert Scott)
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disorder, with an estimated incidence of 1 per 2,500 live births in Caucasian populations. It is caused by mutations in the cystic fibrosis trans-membrane conductance regulator (CFTR) gene which is located on chromosome 7 and encodes for the CFTR chloride channel on cell membranes. Mutations affect CFTR function by a variety of routes including complete loss of the protein, rapid breakdown of CFTR and surface expression of a dysfunctional protein. The most common mutation is ΔF508; a deletion of three nucleotides which results in loss of phenylalanine at the 508th position on the protein causing abnormal tertiary structure and rapid degradation with no apical membrane expression. A very large number of mutations (>500) have been identified in the CFTR gene, but a select group are more commonly associated with disease (e.g. G542X, G551D, N1303K). The prevalence of mutations differs between ethnic populations.
Abnormalities in CFTR cause abnormal ion transport regulation across the cell membrane. In the lungs this results in abnormal airway fluid. Dehydrated mucus is characteristically highly viscoelastic, and adheres to the cilia and airway cells, causing airway plugging. The reduced-volume periciliary fluid layer does not adequately support and lubricate the cilia, and results in defects of ciliary function (Fig. 4.1). Adherent mucus and impaired ciliary function both contribute to reduced airway clearance, chronic infection and biofilm formation. Eventually the chronic infection and inflammation lead to bronchiectasis, which can develop very early in life [23]. Bronchiectasis in CF predominantly affects the upper lobes initially, spreading to all lobes over time. The reasons for upper lobe predominance have been suggested to include aspiration, poor clearance by cough and poor lymphatic clearance to this region, although the disparity with PCD which tends to have worse disease in the middle lobe is difficult to explain.
Although respiratory disease accounts for the vast majority of morbidity and mortality [24]. CF is a multisystem disorder, with manifestations including meconium ileus, pancreatic insufficiency, steatorrhea, failure to thrive, liver disease, diabetes, nasal polyposis and sinusitis. Most cases present before the age of 2 years although later presentation, including in adulthood, is not unheard of particularly in individuals carrying mutations other than ΔF508 who are more likely to be pancreatic sufficient and less likely to have diabetes or pseudomonas colonisation [25]. Diagnosis of classical cases of CF is based on an abnormal sweat test (sweat chloride > 60 mmol/L). Supplementary investigations include CFTR mutations and abnormal transepithelial potential difference.
The majority of patients with CF are easy to diagnose early as they have classical lung disease ± pancreatic insufficiency associated with two CF associated CFTR mutations and an abnormal sweat test result (>60 mmol/l). A further group of patients with mild lung disease and pancreatic sufficiency is confidently diagnosed, often in adulthood, on the basis of a diagnostic sweat test and two disease causing mutations. The group that cause particular difficulty are those with clinically milder phenotypes associated with a variety of equivocal outcomes from genotype and functional investigations (sweat test and nasal potential difference). This is a difficult and controversial area for clinicians and recently international consensus guidelines have been proposed to help in the diagnosis of patients with “CFTR dysfunction that does not fulfil diagnostic criteria for CF” [26]. These single organ disease states are becoming recognised as a spectrum of CFTR related disorders. Very many mutations have been identified in CFTR, but not all are associated with disease and it has been suggested that some only cause disease when interacting with certain environmental factors or other genes. Some patients present with two known disease-causing mutations, but an equivocal sweat test result. These patients generally have more severe lung disease than those with an equivocal sweat test and normal genotype, but they have milder disease than a patient with the same genotype and sweat chloride concentration >60 mmol/l. This highlights different phenotypes associated with a spectrum of test results, indicating the need to perform functional tests even in the presence of two mutations. It is probably appropriate to consider patients with mild CF-like disease associated with two mutations and a normal sweat test as an atypical CF variant. Another group that cause diagnostic uncertainty are those with only one CFTR mutation. These patients may have atypical CF. However the diagnosis of mild or atypical CF with equivocal CF results should only be made after all other causes of bronchiectasis have been excluded. It is prudent to keep the diagnosis under review as our understanding of CFTR related disorders evolves.
Primary Ciliary Dyskinesia
After CF, primary ciliary dyskinesia (PCD) is the most prevalent (1:10–40,000) genetically determined cause of impaired mucociliary clearance. PCD is inherited in an autosomal recessive pattern, and is characterised by chronic infection of the upper and lower airway [21, 27]. The impaired mucociliary clearance is a consequence of abnormal ciliary beat function which is usually [27], but not always [28, 29], associated with abnormal ciliary ultrastructure seen by transmission electron microscopy (Fig. 4.2a–d). Motile cilia are important in organ systems besides the respiratory tract, such as the embryonic node, sperm flagella (Fig. 4.2e), the female reproductive tract, and ependyma of the brain and spinal cord. PCD patients therefore often have extra-pulmonary symptoms caused by dysmotile cilia such as serous otitis media, infertility and rarely hydrocephalus. The cilia of the embryonic node, responsible for left-right asymmetry, are similar in structure to respiratory cilia. Embryonic node dysfunction in PCD causes situs inversus (termed Kartagener’s syndrome) in 50 % of cases (Fig. 4.3a, b) and is associated with congenital heart disease in approximately 6 % of cases [30]. Neonates typically present with respiratory distress of unknown cause and rhinitis. As infants, patients have a persistent wet cough, recurrent respiratory tract infections, rhinitis, and frequently glue ear associated with conductive hearing difficulty. Patients frequently develop sinusitis (Fig. 4.3c). These symptoms may not always be recognised as indicative of PCD and although the mean age of diagnosis is approximately 4 years [31], the range is considerable and reflects local expertise and clinical suspicion. Respiratory symptoms continue into later childhood and adulthood, with many patients developing bronchiectasis, commonly affecting the middle and lower, rather than upper lobes as in CF.
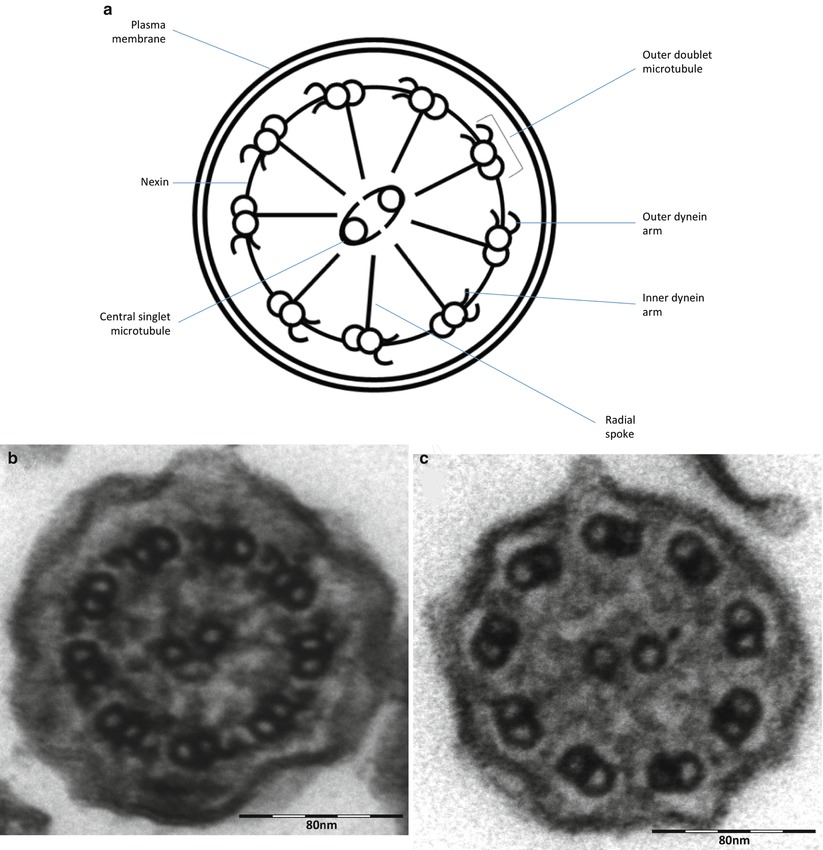
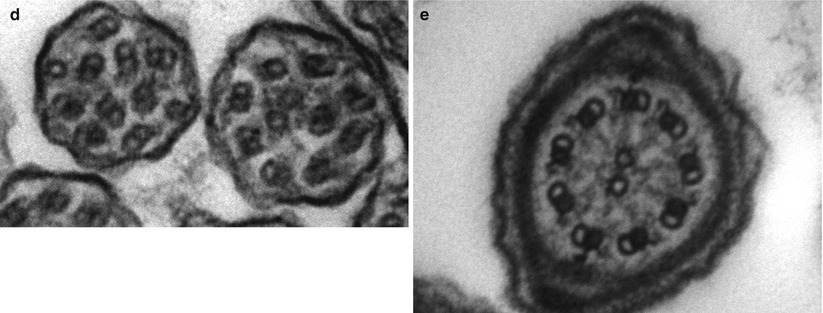
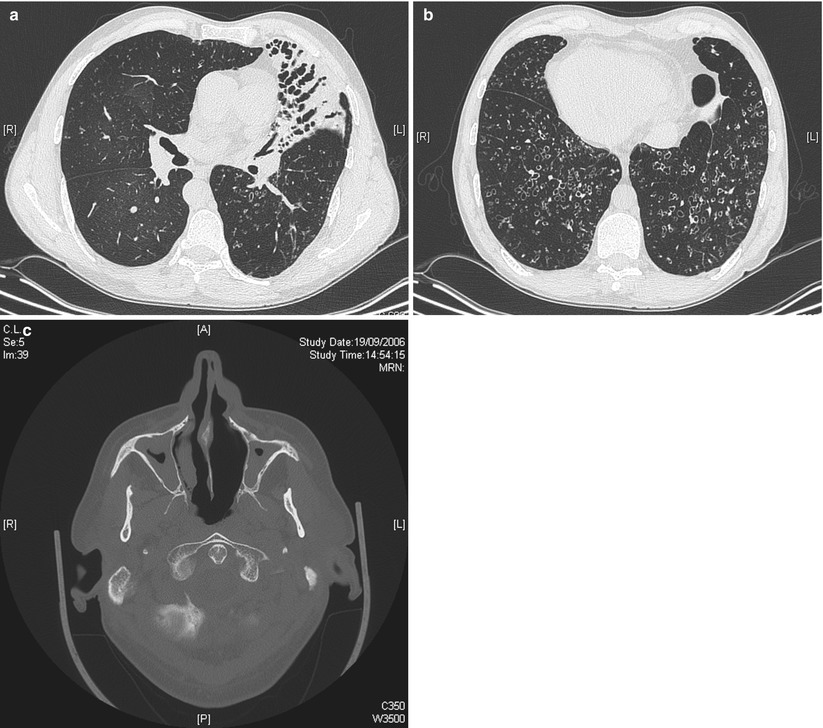
Fig. 4.2
(a) Diagram of transverse section of a respiratory cilium as seen by transmission EM. Motile cilia in the respiratory tract and fallopian tubes have a highly organized “9 + 2” arrangement with nine peripheral microtubule doublets surrounding a central pair of single microtubules running the length of the ciliary axoneme. Nexin and radial spokes maintain the organized structure. Attached to the peripheral microtubules are inner and outer dynein arms. Dynein is a mechanochemical ATPase and generates the force for ciliary beating, hence abnormalities of the dynein arms affect ciliary beating. Transmission EM of a respiratory cilia from (b) a healthy individual and patients with PCD due to (c) an outer dynein arm defect and (d) a radial spoke defect. (e) TEM of a sperm demonstrates similar “9 + 2” ultra-structure (Cartoon image provided by Robert Scott; EM images obtained using FEI Tecnai 12 transmission electron microscope (FEI UK Limited, Cambridge, UK) at 80 kV). Scale bars 580 nm. EM images provided by P. Goggin (Primary Ciliary Dyskinesia Group, University Hospitals Southampton NHS Foundation Trust, Southampton, UK)
Fig. 4.3
HRCT of the chest in a 49-year old man with primary ciliary dyskinesia and Kartagener syndrome at the time of first evaluation for lung transplantation, demonstrating numerous bronchiectases and consolidation in the anterior lateral segment of the left lower lobe (a), and bilateral bronchietasis in the lung bases (associated with centrilobular nodules and tree-in-bud pattern suggestive of bronchiolitis). Note the presence of situs inversus (b). Pansinusitis was present in the same patient (c) (Courtesy of Pr V. Cottin, University of Lyon, France)
The diagnosis of PCD requires specialist investigation, and a number of different tests should be available, since no one investigation can be considered confirmatory [27, 32].
Extremely low levels of nasal nitric oxide are supportive of the diagnosis of PCD [33]. However, some patients with PCD have been described with normal nitric oxide levels, and levels can be low in other conditions including CF. Nasal nitric oxide measurement should therefore only be used as a screening test, and if clinical suspicion is high, further diagnostic investigation should be conducted even if the levels are normal [21].
Patients should have their respiratory epithelial cells visualised by high speed video microscopy for evidence of ciliary dysfunction (e.g. static, dyskinetic, or vibratory cilia). Subtle abnormalities may be significant, but difficult to recognise; facilities should therefore be based at centres with expertise and a high throughput of PCD diagnostic patients. Following abnormal high speed video microscopy analysis, confirmation of diagnosis by an additional method is required because ciliary dysfunction can be secondary, for example to infection. Most, patients with PCD have diagnostic abnormalities of ciliary ultrastructure on transmission electron microscopy and this used to be considered the ‘gold standard’ investigation for PCD (Fig. 4.2). However, it is now recognised that at least 15 % of patients with PCD have normal ciliary ultrastructure and this investigation should not be used in isolation. Transmission electron microscopy analysis requires expert interpretation.
Air liquid interface cell culture techniques are particularly helpful where secondary ciliary defects are suspected, for example due to chronic infection. Re-differentiation of basal epithelial cells to ciliated cells is achieved by culturing the cells, allowing a repeat HSV analysis and electron microscopy having reduced environmental factors that compromise ciliary function for example pollution, infection. However, air liquid interface culture is technically demanding and time consuming; it is routinely available in a small number of specialist centres.
Immunofluorescence microscopy analysis, is a promising technique to help in the clinical diagnosis; it is based on the ability to detect and localise intra-ciliary proteins e.g. DNAH5 by immunofluorescence. However, this method is only routinely available at one centre which is based in Germany.
Genetic testing is currently only able to identify perhaps 50 % of PCD cases, but it is an active area of international research with potential for significant advances. Whilst CF is caused by mutations in one gene, PCD is polygenic with over 200 proteins involved in the formation of cilia. Additionally the disease-causing genes are large, making genetic diagnosis of PCD a challenge. Mutations in 24 genes have been associated with PCD to date, mainly encoding for dynein arm components (reviewed in [32, 34], and summarised in Table 4.2). The two commonest mutations, dynein, axonemal, heavy chain 5 (DNAH5) mutation [37] and dynein arm intermediate chain 1 (DNAI1) mutation [35], have a combined prevalence of 17–35 % in patients with PCD and are associated with static cilia on light microscopy, and dynein arm deficiencies on electron microscopy. Other known mutations are all rare. Mutations in the kintoun gene (KTU) [43], which is required for cytoplasmic pre-assembly of axonemal dyneins results in a similar ciliary phenotype to DNAH5. Mutations in the radial spoke head genes RSPH9 and RSPH4A have been reported in PCD patients with abnormalities of the central microtubular pair causing an abnormal rotating movement of the cilia [56]. Patients with mutations in the dynein axonemal heavy chain 11 (DNAH11) have hyperkinetic vibratory cilia, but apparently normal ultrastructure on electron microscopy [39]. There are therefore a variety of ciliary phenotypes seen on microscopy depending on the responsible mutations, but all result in a severe mucociliary clearance impairment, and clinical phenotype is indistinguishable.
Table 4.2
The genes implicated in PCD
Gene | Ultrastructure defect on electron microscopy | Comments |
|---|---|---|
Genes encoding for outer dynein arm proteins | ||
dynein axonemal intermediate chain 1 (DNAI1) [35] | ODA defect (± IDA) | 2–9 % of all PCD, 4–13 % of PCD with ODA defects [36] |
dynein axonemal heavy chain 5 (DNAH5) [37] | ODA defect (± IDA) | 15–21 % of all PCD, 27–38 % of PCD with ODA defects [38] |
dynein axonemal heavy chain 11 (DNAH11) [39] | Normal | |
dynein axonemal intermediate chain 2 (DNAI2) [40] | ODA defect | Rare (<2 % of cases of PCD) |
thioredoxin domain containing 3 (TXNDC3) [41] | ODA defect | Rare (<2 %) |
DNAL1 [42] encoding the ODA light chain1 | ODA | |
Genes encoding for assembly, transport or attachment of proteins | ||
kintoun (KTU)/ dynein, axonemal, assembly factor 2 (DNAAF2) [43] | ODA & IDA defects | 12 % of PCD with ODA + IDA defects [36] ktu encodes for a cytoplasmic protein responsible for pre-assembly of dynein arm complexes in the cytoplasm |
Leucine rich repeat containing 50 (LRRC50) [44] | ODA & IDA defects | Rare |
Dynein, axonemal, assembly factor 3 ( DNAAF3) [45] | ODA & IDA defects | |
ODA & IDA defects | 11 % of PCD with ODA + IDA defects | |
Coiled-coil domain containing 103 (CCDC103) [48] | IDA and ODA defect | CCDC103 acts as a dynein arm attachment factor. Mutations disrupt assembly of dynein arms |
ODA defect | CCDC114 is required for attachment of ODAs in the axoneme. 6 % of PCD with ODA defects [36] | |
HEAT repeat containing 2 (HEATR2) [52] | ODA absent | HEATR2 localises to cytoplasm suggesting a role in dynein arm transport or assembly |
Coiled-coil domain containing 65 (CCDC65) [53] | Normal EM, vibrating cilia | Nexin-dynein regulatory complex component absent from airway cells of patient |
ODA and IDA absent | Gene is essential for axonemal assembly of dynein arms | |
Genes encoding for radial spoke head proteins and genes associated with central pair abnormalities | ||
radial spoke head protein 9 (RSPH9) [56] | Intermittent or complete central pair defect | |
RSPH4A [56] | ||
RSPH1 [57] | Central complex and radial spoke defect | |
HYDIN [58] | cilia lack the C2b projection of the central pair apparatus | |
Coiled-coil domain-containing protein 39 (CCDC39) [59] | Axonemal disorganisation and IDA defect (sometimes called ‘radial spoke defect’ | 36–65 % of PCD with IDA defects + axonemal disorganization. CCDC40 is required for the axonemal localization of CCDC39. The mechanisms mediated by these proteins are unclear |
CCDC40 [60] | Axonemal disorganisation and IDA defect | 24–54 % of PCD with IDA defects + axonemal disorganization [36] |
Defect of nexin-dynein regulatory complex | ||
DRC1 CCDC164 [61] | Axonemal disorganization in small proportion of cilia | Absent nexin-dynein regulatory complex (N-DRC) |
Associations with other syndromes | ||
Oral-facial-digital type 1 (OFD1) [62] | Unknown | One case report |
Retinitis Pigmentosa guanosine triphosphatase regulator (RPGR) [63] | Variable | Association with X linked retinitis pigmentosa |
In extremely rare cases, PCD has been genetically linked to other ciliopathies. X-linked recessive retinitis pigmentosa, sensory hearing deficits, and PCD have been associated with mutations in the Retinitis Pigmentosa guanosine triphosphatase regulator (RPGR) [63]. A single family has been reported with a novel syndrome that is caused by Oral-facial-digital type 1 syndrome (OFD1) gene mutations characterized by X-linked recessive mental retardation, macrocephaly, and PCD [62].
Young’s Syndrome
Young’s syndrome, or Barry-Perkins-Young syndrome, is a clinical entity characterised by bronchiectasis, chronic sinusitis and impaired fertility. The diagnosis is considered most commonly in middle aged men presenting with infertility. Although Young’s syndrome has been reported in identical twins prompting suggestions of a genetic aetiology, there is some evidence that this syndrome has declined following the removal of mercury from teething powders and worm medicines in the 1950s [64]. Moreover, whilst Young’s syndrome and other disorders of mucociliary clearance have partially overlapping symptomatology, a common causative link has not been identified. Indeed the mechanism of reduced fertility in Young’s appears to be due to functional obstruction of sperm transport down the epididymal tract rather than due to absence of the vas deferens which is seen in CF or to dysmotility seen in PCD. There are reports that mucociliary clearance is impaired in Young’s syndrome [65] but ciliary ultrastructure is largely normal [66]. Similarly, patients with Young’s are not frequently carriers of common CF mutations [67], although possibly there may be an association with atypical CF mutations [68]. Some authors have even questioned whether Young’s syndrome exists as a recognised clinical entity at all [69], and it is quite possible that as diagnoses of rare variants of CF are established, Young’s syndrome will disappear as a diagnosis.
Other Ciliopathies
Non-motile or ‘primary’ cilia are found on the surface of many cells in the body. An increasing number of diseases are attributed to abnormal motile or primary ciliary function, collectively known as ciliopathies (http://www.ciliopathyalliance.org). For example, in the eye and kidney ciliopathies can cause retinitis pigmentosa, autosomal dominant polycystic kidney disease (ADPKD) or nephronophthisis. Primary cilia are similar in structure to the respiratory epithelial cilia (9 + 2), but in cross section, there is no central pair of microtubules (9 + 0). There are several case reports of patients with PCD-like disease in association with retinitis pigmentosa [70, 71]. In some patients it may be that both primary and motile cilia are impaired. In others it may be that the problem is purely of primary cilia, with abnormalities of primary pulmonary cilia causing lung disease. This needs further evaluation.
Autosomal dominant polycystic kidney disease (ADPKD) is an example of renal ciliopathy associated with bronchiectasis. It affects between 1 in 400 and 1 in 1,000 people [72]. The disease most commonly manifests in adulthood and is caused by defective ciliary function in renal epithelial cells. Two genes, PKD1 and PKD2 coding for proteins known as polycystins have been implicated in the pathogenesis of ADPKD. In ADPKD, impaired primary cilial sensing results in abnormal intracellular signalling, cell hyperproliferation, and cyst formation [73]. A predisposition to bronchiectasis is recognised, although the mechanism for this is not fully understood [74].
Alpha-1 Antitrypsin Deficiency
An association between alpha-1 antitrypsin (AAT) deficiency and emphysema is well-known but there are also reported cases of an association with severe PiZ genotypes and bronchiectasis, this is largely seen in adulthood. A study of 74 patients with severe AAT deficiency found high resolution CT scan evidence of bronchiectasis in 70 subjects and judged this to be clinically significant in 20 [75]. AAT deficiency alleles are over represented in patients with bronchiectasis and asthma combined, suggesting that bronchiectasis may occur as a consequence of airway obstruction, in turn reducing airway clearance [76].
Disorders of Immunity
Immunodeficiency accounts for a significant proportion of cases of bronchiectasis in developed countries. Recent series suggest as many as 7 % of adults [77] and 20–30 % of paediatric cases in the developed world may be attributable to primary immunodeficiency [1, 10]. Immunodeficiency should be considered following respiratory infections that are unusually severe, recurrent, unresponsive to conventional treatment, or atypical [78]. Common associated features include failure to thrive, severe atopic disease, and occasionally, auto-immune disease [79]. Primary immunodeficiencies include a heterogeneous group of disorders of immune development or function affecting innate or adaptive immunity. Common variable immunodeficiency (CVID), X-linked agammaglobulinemia (XLA) and chronic granulomatous disease (CGD) are the most common immunodeficiencies found in association with bronchiectasis [1, 10]. Disorders of innate immunity are currently poorly characterised but are likely to be responsible for a proportion of cases of bronchiectasis currently labelled ‘idiopathic’.
Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) has an estimated prevalence of 1 per 25,000 population and is the commonest immunodeficiency. Presentation is usually in young adulthood but diagnosis may be delayed [80]. Patients affected by CVID display a defective antibody response to protein and polysaccharide antigens and low IgG, IgM and IgA levels. This places affected individuals at risk of recurrent bacterial infection as well as autoimmune disease and malignancy [81]. Clinical features vary in CVID, perhaps reflecting heterogeneity of the molecular defects underlying the disease. Susceptibility loci for CVID have been identified within the MHC class II alleles [82], and polymorphisms in the tumour necrosis factor gene have been also been reported in association with CVID [83]. Mutations associated with CVID have been identified within the inducible costimulator (a molecule expressed by T cells) [84], CD19 (a T cell marker important in B cell development, activation and proliferation) [85], and in transmembrane activator and CAML interactor (a receptor believed to be important in antibody responses to type II T-independent antigens) [86].
In individuals with CVID at least one episode of pneumonia has usually occurred before diagnosis of CVID is made [81]. Possibly as a consequence of delayed diagnosis, the risk of bronchiectasis is greater in patients with CVID than in those with X-linked agammaglobulinemia or other immunodeficiency diseases and may approach 70 % [87]. Bronchiectasis tends to be more common in the lingual, middle and lower lobes. The threshold for HRCT imaging should be low in cases of known or suspected immunodeficiency, particularly if there are signs of persisting lung disease [78].
X-Linked Agammaglobuinemia
X-linked agammaglobuinemia (XLA) is characterised by almost complete absence of circulating B lymphocytes and of all immunoglobulin [88]. Mutations of the Bruton’s tyrosine kinase gene cause an incomplete block of B cell development at the pre-B cell stage [89]. This leaves affected patients susceptible to infection by encapsulated bacteria and mycoplasma [90]. More recently autosomal recessive forms of agammaglobulinemia have been identified that are caused by mutations in genes that encode for other components of the pre-B cell receptor and its signalling pathway [91]. Pulmonary infections can occur shortly after birth but generally become noticeable beyond 6 months of age following the disappearance of maternal IgG. The most common age for diagnosis is under a year but presentation as late as 5 years has been known [92]. Although a recent survey has found less than a third of adults with XLA to be severely affected by bronchiectasis [93], there is evidence to suggest that the risk of developing significant lung disease increases over time and can be reduced by early detection and treatment [94]. Chronic lung disease, including bronchiectasis, has also been observed in children with IgA deficiency, particularly when associated with IgG2 deficiency [95].
Chronic Granulomatous Disease and Other Disorders of Neutrophil Function
Chronic granulomatous disease (CGD) results from impaired function of NADPH oxidase. This enzyme is required for effective functioning of the phagocytic respiratory burst and for superoxide production. Impaired NADPH oxidase is generally transmitted by X-linked inheritance but autosomal recessive variants are also recognised [96]. Mean age at presentation in autosomal recessive disease is 10 years, slightly later than X-linked disease where the mean is 5 years suggestive of a more severe phenotype [97]. Sufferers are vulnerable to recurrent and severe bacterial and fungal infections, frequently Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia and Aspergillus spp.
A similar pattern of recurrent pneumonia and lung aspergillosis may also be observed in patients with severe congenital neutropenia [98]. Most commonly this disorder occurs as a consequence of mutations in the gene encoding for neutrophil elastase [99] but can also be attributable to mutations affecting a mitochondrial protein thought to be involved in protecting myeloid cells from apoptosis [100] or an endosomal protein involved in intracellular signalling [101]. The characteristic feature of this disease is low levels of circulating neutrophils and hence vulnerability to bacterial and fungal pathogens.
Bronchiectasis, alongside severe pneumonias, empyemas and pneumatoceles, is common in children with hyper-IgE or Job’s syndrome, in which neutrophil defects occur in combination with a wide variety of lymphocyte and humoral function defects as well as very high levels of serum IgE [102]. Job’s syndrome is inherited in an autosomal dominant pattern. Mutation of the signal transducer and activator of transcription 3 or STAT3 gene is known to cause Job’s syndrome [103], although in some cases the responsible mutation is unknown. STAT3 is a transcription factor which influences the expression of a variety of genes and plays a key role in many cellular processes such as growth and apoptosis. An autosomal recessive form of Job’s syndrome is recognised, this is less common than the autosomal dominant form and less likely to have respiratory complications. Bronchiectasis is also seen in association with Shwachman-Bodian-Diamond syndrome, a rare autosomal recessive disorder characterised by exocrine pancreatic insufficiency, bone marrow dysfunction, leukemia predisposition, and skeletal abnormalities. In most cases, Shwachman-Bodian-Diamond syndrome is associated with mutations in the Shwachman-Bodian-Diamond Syndrome (SBDS) gene located on chromosome 7 [104]. Bone marrow dysfunction results in neutropenia in the majority of patients and may be accompanied by defects in neutrophil mobility, migration, and chemotaxis.
Other Immunodeficiency Diseases Associated with Bronchiectasis
It is currently difficult to identify many causes of innate immunodeficiency, and it is likely that as new defects are discovered, many cases currently labelled as suffering from ‘idiopathic bronchiectasis’ will have an underlying cause. Rare instances of bronchiectasis in association with deficiency of C2, or mannose binding lectin, have been reported and an association between deficiency of l-ficolin and bronchiectasis has been reported in adult patients [105]. Complement deficiency is also known to affect the severity of bronchiectatic disease in CVID [106] and in CF [107].
Ataxia telangiectasia is an autosomal recessive multisystem disorder resulting from mutation of the Ataxia telangiectasia mutated (ATM) gene, characterised by the development of telangiectasia and cerebellar ataxia. It is the most common of the DNA repair disorders and is associated with chromosomal instability and cellular radiosensitivity rendering sufferers susceptible to cancer and to infection. The ATM gene is involved in antibody class switch recombination and defects in this process may underlie the increased susceptibility of ataxia telangiectasia patients to bacterial infections [108]. The most common humoral immunological defects are diminished or absent serum IgA and IgG2, and impaired antibody responses to vaccines [109]. Ataxia telangiectasia leads to thymic hypoplasia and variable T cell deficiency. It is likely that recurrent aspiration due to swallowing impairment also contributes to respiratory disease [110]. Fifty percent of patients die in adolescence from overwhelming bronchopulmonary disease [109].
Collagen Disorders
Although the majority of causes of bronchiectasis are related to defects of mucociliary clearance or immunodeficiency, congenital abnormalities affecting the structure of the bronchial wall can predispose to bronchiectasis. In particular, a number of congenital syndromes of collagen and cartilage abnormalities have been associated with bronchiectasis. This suggests that abnormally compliant or distended bronchi can predispose to bronchiectasis.
Marfan Syndrome
Marfan syndrome is a rare hereditary disorder characterised by skeletal, cardiovascular and ocular abnormalities. Pulmonary abnormalities occur in approximately 10 % of patients, the commonest being spontaneous pneumothorax and emphysema [111]. Although rare, cases of bronchiectasis in adults [112] and children [113] who have Marfan syndrome have been described. Marfan syndrome is autosomal dominantly inherited with variable expression; features of the condition arise as a consequence of a defect in Type 1 collagen. The defect in collagen may be responsible for the reduced tensile strength of the connective tissue leading to bronchiectasis and increased susceptibility to infection.
Other Collagen Disorders
In the 1960s Williams and Campbell first described a series of children with bronchiectasis who had a bronchial cartilage deficiency from the third division with normal trachea and central bronchi [114]. It is hypothesised that the compliant bronchi collapse during coughing, leading to poor airway drainage [115]. The familial pattern and the early onset of symptoms support the possibility that this is a rare congenital syndrome [116]. Williams-Campbell syndrome has most frequently been described in children although adult cases have been reported. CT imaging demonstrates bilateral bronchiectasis affecting segmental and subsegmental bronchi [117]. Cartilage deficiency is not evident outside of the bronchi and the cause of the deficiency is not well understood. It has been hypothesised that children who are born with a bronchial cartilaginous defect develop pathology after suffering a viral respiratory infection in infancy. As a precise genetic defect has not been identified and solitary cases have been reported with greater frequency than familial, the genetic basis of Williams-Campbell syndrome is debateable, indeed secondary cartilaginous damage due to infection and inflammatory change cannot be completely excluded.
A contrasting condition characterised by dilated trachea and large bronchi was first described by Mounier-Kuhn in 1932 [118]. This idiopathic tracheobronchomegaly is associated with varying degrees of bronchiectasis varies in age and severity at presentation but usually presents in mid-adulthood. Males are more frequently affected than females, and a racial predominance has been proposed [119]. The trachea may have a ridged appearance with multiple diverticula. Dynamic collapse of the enlarged airways in expiration, particularly posteriorly, is also likely [120] and pooling of secretions may predispose to lower respiratory tract infections [118]. The aetiology of Mounier-Kuhn syndrome remains uncertain, although histological data suggest that tracheal and bronchial dilatation occur as a consequence of abnormal development of airway connective tissue, with a deficiency of elastic and muscular fibres [121]. Tracheobronchomegaly occurs in association with other congenital connective tissue disorders, including Ehlers Danlos syndrome [122] and cutis laxa [123]. Together with reports of the disorder occurring in siblings [119] this makes an unidentified genetic cause possible, at least in some cases. Generally, there is no curative treatment for the syndrome, although the use of tracheobronchial prostheses may improve symptoms in adults [124].
Other Genetic Predispositions to Bronchiectasis
Bronchiectasis has been reported in association with autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, relapsing polychondritis and inflammatory bowel disease [125]. A genetic association in ulcerative colitis-associated bronchiectasis has recently been reported with functional polymorphisms in the cytokine IFNγ and the neutrophil chemokine, CXCR1 [126]. Yellow nail syndrome is a rare disorder observed in association with a number of systemic diseases including rheumatoid arthritis, and also reported in cases of tuberculosis, immunological disorders and malignancies [127]. This syndrome is characterised clinically by dystrophic yellow nails, lymphoedema and pleural effusion [128]. A significant number of patients also have sinusitis [129], lower respiratory tract infections [130], and bronchiectasis [131]. The syndrome is most often seen in middle-aged individuals although a case report has described bronchiectasis in a 6-year-old [132]. Congenital lymphatic malformation and dysfunction is believed to be responsible for the syndrome [133], although a selective deficiency of humoral immune function has also been suggested [134]. Whilst the exact aetiology is unknown, a genetic component has been proposed [135].
Idiopathic Bronchiectasis
An underlying cause cannot be found in at least 30 % of patients with bronchiectasis, these cases are referred to as idiopathic [1, 10, 14, 77]. Some cases are familial and it is likely that a number of patients have unrecognised impairment of the innate immune system, or have one of the increasingly recognised CFTR mutations associated with milder CF phenotypes causing isolated lung disease. CFTR mutations have been found to be overrepresented in individuals identified as suffering from idiopathic bronchiectasis who do not have a full CF phenotype. The 5 T CFTR mutation in particular has been found at high frequency in this patient group. Recent data suggest that the 5 T polythymidine tract sequence of intron 8 on specific haplotype backgrounds (TG12 and M470V) may underlie low levels of full-length functional CFTR protein and cause CF-like lung disease [136].
Diagnosis
A number of international guidelines exist for the diagnosis and management of bronchiectasis in adults and children [15, 78, 137]. Ideally patients at risk of bronchiectasis should be identified before irreversible damage develops. For example, it is widely accepted that newborn screening for CF, with early introduction of prophylactic treatment, and aggressive management of infections, reduces long-term pulmonary morbidity [138–140]. Similarly in PCD, observational data suggests that lung function decline is stabilised and can even be reversed following diagnosis and instigation of appropriate pulmonary management [141–143].
A diagnostic approach to a patient with symptoms suggestive of bronchiectasis is summarised in Fig. 4.4. HRCT is the gold standard investigation for bronchiectasis, but a number of non-specific investigations assist evaluation of disease severity. A history of recurrent or prolonged wet cough is suggestive of significant endobronchial infection which may progress to bronchiectasis. Individuals in whom bronchiectasis should be suspected include those with a persistent cough for at least 8 weeks [78]. Intermittent hemoptysis is common, particularly in adults. Finger clubbing and persistent wet crackles are indicative of severe disease.
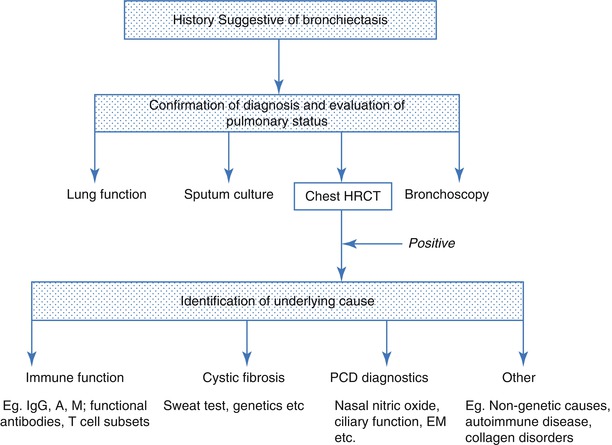
Fig. 4.4
A diagnostic approach to bronchiectasis
Investigation of patients with prolonged lower respiratory tract symptoms should usually include a standard posterolateral chest x-ray and culture of sputum. Dilated airways with thickened walls are sometimes visible either on chest x-ray as parallel ‘tram tracks’ or ‘ring shadows’. Fluid-filled bronchi may be visible as ‘gloved-finger’ shadows. Situs inversus might direct investigations for PCD (Fig. 4.5). However chest x-ray is not a sensitive test for bronchiectasis, and if the diagnosis is suspected, further investigation is warranted. Patients should have sputum cultured; Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, non–tuberculous mycobacteria or Burkholderia cepacia are suggestive of significant lower respiratory tract pathology [78]. Measures of lung function are non-specific and non-sensitive in bronchiectasis, but contribute to the assessment of disease severity. Forced expiratory volume in 1 s (FEV1) is often normal in early disease although a reduced FEV1 in the presence of normal functional vital capacity (FVC) is common. Standard spirometry is not a good measure of disease decline, and alternative measures are sought. Lung clearance index is a measure that has been suggested to be a good monitor of disease in CF [144] and is being evaluated as an early marker of lung disease in other bronchiectatic diseases such as PCD [145].
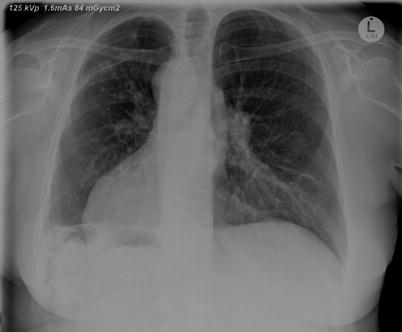
Fig. 4.5
A chest xray of an adult with PCD associated with dextrocardia. There is increased bronchial wall thickening, and also blunting of the right costophrenic angle. The patient had previously had a lung resection via a thoracotomy on the right for bronchiectasis
High-resolution computed tomography (HRCT) has sensitivity and specificity in excess of 90 % [146] and may detect signs of bronchiectasis not identified by plain film imaging [147]. For this reason HRCT is the gold standard diagnostic investigation (Figs. 4.6a, b and 4.7). Diagnostic radiological criteria were described by Naidich et al. in 1982 [148] and are also summarised in the Fleischner Society: glossary of terms for thoracic imaging [149]. Characteristically enlarged bronchi seen in cross-section are larger than the accompanying arteries giving rise to the ‘signet ring’ sign. Other characteristic features include dilated airways with air-fluid levels that do not taper and remain visible in the extreme lung periphery [150]. Expiratory HRCT images can be useful. Parenchymal hypoattenuation may identify small airways disease due to air-trapping associated with mucous-filled bronchi in bronchiectasis and evidence of bronchial wall or tracheal collapse can also be detected, providing an alternative investigation to bronchoscopy [151]. HRCT should not be performed during acute respiratory exacerbations as bronchial dilation is difficult to assess in the presence of consolidated lung, whilst pulmonary collapse can cause misleading ‘traction bronchiectasis’ by pulling on neighbouring bronchi [152].
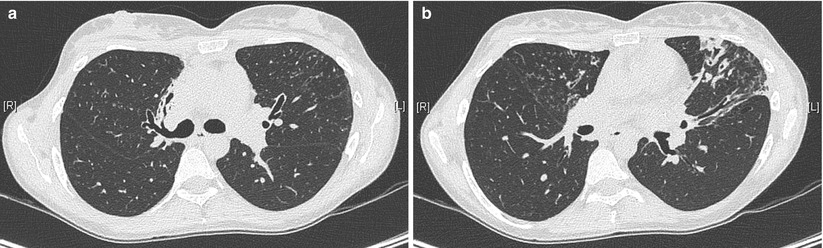
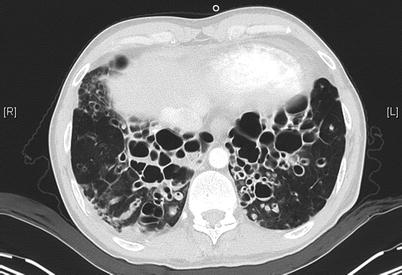
Fig. 4.6
HRCT of the chest in a 32-year old woman with primary ciliary dyskinesia, demonstrating bronchiectasis in the right middle lobe (a) and the left upper lobe (b). Centrilobular nodules suggest associated bronchiolitis (Courtesy of Pr V. Cottin, University of Lyon, France)
Fig. 4.7
HRCT of the chest in a 47-year old man with primary ciliary dyskinesia evaluated for lung transplantation, demonstrating prominent bilateral bronchiectases in the lower zones of the lungs (Courtesy of Pr V. Cottin, University of Lyon, France)
Fibreoptic bronchoscopy should be considered in patients with a prolonged wet cough to assess airway structure and calibre and exclude pathology such as severe tracheomalacia, bronchomalacia or tracheal bronchi which may contribute to bronchiectatic change. Bronchoscopic examination can also provide lavage fluid for evidence of chronic aspiration measured by pepsin, amylase or fat-laden macrophages, and for culture and microscopy.
Once a diagnosis of bronchiectasis has been made, investigation of the underlying cause should be sought. The clinical history should direct investigations which will usually include investigation for CF, PCD and immunodeficiency (Table 4.3).
Table 4.3
Characteristic features and diagnostic criteria for bronchiectasis of known genetic origin
Diagnosis | Genetic basis | Suggestive symptoms | Age at diagnosis | Diagnostic criteria |
|---|---|---|---|---|
Cystic fibrosis | Mutation of CFTR gene 7q31-7q32 | Meconium ileus | Birth if discovered by screening, otherwise presentation is generally in infancy due to failure to thrive or recurrent respiratory infection. Approximately 10 % of cases present as adults. | Immunoreactive trypsin newborn screening |
Most commonly the ΔF508 mutation | Rectal prolapse | Sweat chloride testing | ||
Failure to thrive | Genotype analysis | |||
Steatorrhea | CF Trust guidelines [153] | |||
Nasal polyps | ||||
Chronic sinusitis | ||||
Male infertility | ||||
Primary ciliary dyskinesia | Polygenic disorder affecting cilial structural proteins [32] | Neonatal tachypnoea | Although symptoms may be present in the neonatal period these are often identified retrospectively. Diagnosis can be at any stage of life although mean age of diagnosis in childhood is 4 years, and many adults remain undiagnosed. | Ciliary biopsy for high speed video microscopy, transmission electron microscopy and air liquid interface cell culture techniques |
Chronic otitis media with possible speech delay | Nasal nitric oxide measurement | |||
Chronic rhinitis | ||||
Situs inversus | ||||
Common variable immunodeficiency | Mutations in but not limited to ICOS, TNFRSF13B, TNFRSF13C and CD19 genes | Low immunoglobulin levels | Median age of symptom onset is third decade although may be earlier. Diagnosis is often delayed by 5–10 years from symptom onset. | IgG and IgA and/or IgM |
Recurrent bacterial infections | >2 SD below mean for age | |||
Defective antibody response to protein and polysaccharide antigens | ||||
Diagnostic criteria [155] | ||||
X-linked agammaglobulinaemia | Mutation of Bruton’s tyrosine kinase gene | Male patient | Prenatal diagnosis possible in families where mutation is known. Presentation generally in the first year of life although can be up to 5 years | IgG and IgA and IgM > 2 SD below mean for age |
Onset beyond the first 6 months of life | Absent isohaemagglutinins and/or poor response to vaccines | |||
Low immunoglobulins and absent circulating B cells | Diagnostic criteria [155] | |||
Encapsulated bacteria and mycoplasma infections | ||||
Chronic granulomatous disease | Mutations affecting NADPH oxidase | Recurrent bacterial and fungal infections | Presentation at mean age of 5 years for x-linked forms and 10 years for autosomal recessively inherited disease | Abnormal nitro blue tetrazolinium reduction Respiratory burst <5 % of control in activated neutrophils |
gp91, p22, p47, p67 phox | Liver, perirectal, lung or bone infection | Diagnostic criteria [155] | ||
Failure to thrive | ||||
Hepatosplenomegaly lymphadenopathy | ||||
Marfan syndrome | FBN1 mutations in fibrillin gene on chromosome 15 | Ectopia lentis (displacement of the lens of the eye)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|