Unexplained cardiac arrest (UCA) can be caused by low-penetrance genetic disorders. The aim of this cross-sectional study is to assess the usefulness of a new diagnostic protocol: Thirty-five patients were recruited from 9 Spanish centers. Electrocardiogram, echocardiogram, and coronary catheterization were used to rule out electrical or structural heart disease in all subjects. Patients underwent pharmacologic tests with epinephrine and flecainide, followed by assessment of family members using electrocardiogram and echocardiogram, and next-generation genetic sequencing to analyze 126 genes if all the other test results were negative. A firm diagnosis of channelopathy required phenotypic proof of the condition in unmasking tests, the presence of a pathogenic variant consistent with the phenotype observed, and/or co-segregation of the mutation found in a family member’s phenotype. A firm diagnosis was made in 18 cases. The diagnoses were 7 Brugada syndrome, 5 catecholaminergic polymorphic ventricular tachycardia, 3 long QT syndrome, 2 early repolarization syndrome, and 1 short QT syndrome. Pharmacologic testing was the most frequent method of diagnosis. In 5 cases, the diagnosis was made based on positive genetic testing without phenotypic alterations. In conclusion, this sequential diagnostic protocol allows diagnoses to be made in approximately half of the UCA cases. These diagnoses are low clinical penetrance channelopathies. If interpreted carefully, genetic tests can be a useful tool for diagnosing UCA without a phenotype.
Unexplained cardiac arrest (UCA) is a rare condition caused primarily by low clinical penetrance cardiac channelopathies and cardiomyopathies. The main causes are long QT syndrome (LQTS), short QT syndrome (SQTS), Brugada syndrome (BS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and arrhythmogenic right ventricular cardiomyopathy (ARVC). Identifying the underlying cause of UCA is essential to provide targeted pharmacologic treatment in the proband and above all to identify other potentially treatable cases in family members. This report describes the usefulness of a sequential diagnostic protocol including pharmacologic tests, family assessment, and wide-spectrum genetic testing using next-generation sequencing in a multicenter registry of UCA.
Methods
In this study, UCA was defined as ventricular fibrillation (VF) with no diagnostic findings on the electrocardiogram (ECG), no pathologic findings on the echocardiogram, and no angiographic lesions with >50% stenosis on coronary catheterization. Recording of a VF rhythm was not required to be included, but the needing of an external defibrillation to terminate the episode was an obliged inclusion criteria.
Patients were recruited prospectively and from each center’s retrospective cohort during 3 years, in a cross-sectional design study. The exclusion criteria used were baseline ECG with prolonged QT (QTc interval >460 ms in male and >480 ms in female subjects), Brugada pattern in right precordial leads and/or pathologic Q waves, documented monomorphic ventricular tachycardia, structural heart disease on transthoracic echocardiogram or cardiac magnetic resonance imaging (MRI), probable channelopathy in the proband or first-degree relatives, or ARVC, in accordance with published criteria, evidence of drug use, severe electrolyte abnormality, ischemia, extreme bradycardia, or any other secondary cause of VF and coronary artery anomaly/vasospasm.
During 3 years, 51 patients with apparently UCA were initially recruited. From these, 13 were excluded because of subtle pathologic findings observed in conventional tests and 3 did not consent for pharmacologic tests. Finally, 35 patients from 9 participating centers were included. All subjects signed the informed consent form, and the study was approved by the ethics committee of each participating hospital.
Figure 1 shows the sequential diagnostic protocol undergone by each index case. The ECGs, echocardiograms, and coronary angiograms were exhaustively reviewed to rule out the presence of evident electrical or structural heart disease. After this, all subjects underwent the sequence of diagnostic steps shown in Figure 1 . If all the test results were negative and an early repolarization pattern was seen on the ECG, the case was categorized as early repolarization syndrome in accordance with the latest published data. Early repolarization was defined as J-point elevation (QRS-ST junction) of at least 1 mm in 2 contiguous inferior or lateral leads.
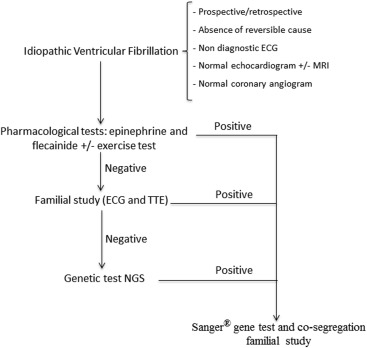
The first step in the diagnostic protocol consisted of the pharmacologic tests with epinephrine and flecainide. Ergometric tests were performed following the standard Bruce protocol if the epinephrine test was contraindicated. The epinephrine test was performed first. If negative, it was followed by the flecainide test after a washout period. The epinephrine test was performed following the Shimizu protocol. In accordance with Shimizu and Khran, the test result was considered positive for LQTS if a prolonged absolute QT interval >30 ms was observed with sinus tachycardia or if the ascending limb of the T wave was notched, indicative of type 2 LQTS. CPVT was diagnosed if there were >3 polymorphic ventricular ectopies or if bidirectional VT developed. Flecainide test result was considered positive if ST-segment elevation of >1 mm was observed in at least 1 standard or high right precordial leads (Brugada type 1 pattern).
If the pharmacologic test results were negative, the subject moved on to step 2 of the diagnostic process as shown in Figure 1 . This diagnostic step consisted of performing ECGs and transthoracic echocardiograms in available first-, second-, and third-degree relatives. First-degree relatives include parents, offspring, and siblings, and the remaining were second- or third-degree relatives. If these test results revealed abnormalities consistent with a channelopathy (borderline QTc interval or Brugada type 2 pattern), pharmacologic unmasking tests were performed.
Genetic testing using next-generation sequencing was performed in all patients where there was no phenotype information in the previous diagnostic steps. The test consisted of mass sequencing of 126 genes linked to the cardiomyopathies and channelopathies described, using non–Sanger-based high-throughput DNA sequencing technologies. These genes included KCNQ1 , KCNH2 , SCN5A , KCNJ2 , RYR2 , CASQ2 , CACNA1B , and desmosome and sarcomere genes (see Supplementary Table 1 ). In patients in whom a diagnosis was made, by whatever means, genetic testing was performed using the conventional DNA sequence method, “Sanger,” targeted at the genes involved in the condition studied. These genes were SCN5A for BS, KCNQ1 , KCNH2 , SCN5A , KCNE1 , KCNE2 for LQTS and RyR2 and CSQ2 for CPVT.
Results
All the 35 patients recruited underwent a baseline ECG, transthoracic echocardiogram, and coronary catheterization, revealing no relevant abnormalities. Cardiac MRI was performed in 13 cases and an electrophysiology study in 15, with no pathologic findings. Participant age ranged from 4 to 66 years (average 39.8 ± 15.7 years) and 21 were men. Four cases had a history of syncope, 6 had a family history of sudden death, and, in most cases, VF was documented during cardiac arrest. Table 1 lists the baseline characteristics of the cohort in relation to whether a final diagnosis was made.
| Diagnosis (n=18) | No diagnosis (n=17) | P-value | |
|---|---|---|---|
| Age (years) | 36.6 ± 18.0 | 42.3 ± 15.6 | 0.30 |
| Male | 64.7 % | 55.5 % | 0.66 |
| Family History of sudden death | 11.7 % | 22.2 % | 0.61 |
| Previous syncope | 17.6 % | 5.5 % | 0.54 |
| ECG | |||
| PR (ms) | 159.6 ± 18.4 | 161.3 ± 25.4 | 0.88 |
| QRS (ms) | 87.0 ± 7.8 | 87.1 ± 8.9 | 0.89 |
| QTc (ms) | 394.9 ± 22.1 | 399.1 ± 28.0 | 0.65 |
| Magnetic Resonance Imaging performed | 27.7 % | 47 % | 0.13 |
Table 2 summarizes the overall results of the diagnostic study. A firm diagnosis was made in 51.4% of cases—16 cases through any of the 3 steps of the algorithm and 2 more with early repolarization syndrome after a negative result in all tests. Of the 17 remaining cases, data suggestive of origin were found in 8 patients, but the definitive diagnostic criteria for the pathology in question were not met, and in 9 cases, the test results were negative.
| Step 1 (pharmacological tests) | Step 2 (family assessment) | Step 3 (genetic tests) | Total | |
|---|---|---|---|---|
| Brugada syndrome | 4 | 3 | 0 | 7 |
| Long QT syndrome | 2 | 1 | 0 | 3 |
| CPVT | 1 | 0 | 4 | 5 |
| Short QT syndrome | 0 | 0 | 1 | 1 |
| Early repolarization syndrome | 0 | 0 | 0 | 2 |
| Diagnosis | 7/35 (20%) | 4/28 (14.2%) | 5/24 (20.8%) | 18/35 (51.4%) |
In 7 cases, the final diagnosis was made in step 1. Four of these diagnoses were of BS, 2 were of LQTS, and 1 was of CPVT. Figure 2 shows 2 examples of diagnoses made based on an epinephrine test. In both cases, the Sanger technique confirmed the presence of a pathogenic mutation in an expected gene ( KCNH2 Pro347Ser in the LQTS case and RyR2 Leu4915Trp in the CPVT case). Interestingly, in all cases with a positive epinephrine test result, the ergometric test results had been negative.
ECGs in family members (step 2) identified the cause in 4 cases. Three were cases of BS where the index case had no abnormalities on the baseline ECG or after flecainide, but a first-degree relative did. In one of these 3 cases, co-segregation with the pathogenic variant p.D1816Vfs in SCN5A was confirmed in 7 carrier relatives ( Figure 3 ). This mutation causes severe truncation (201 residues) of the C-terminus of Nav1.5 and the functional analysis revealed a marked reduction in channel trafficking toward the membrane and in sodium current density compared with nonmutated channels (Wild Type). In another case, despite a negative adrenaline test in the proband, who had severe neurologic damage, an upper limit of normal QTc was detected in a first-degree relative. The test was, therefore, performed in 3 more first-degree relatives who all tested positive for LQTS ( Figure 3 ) and received treatment with β blockers.
Subclinical channelopathy was diagnosed in 5 cases based on the detection of a genetic variant. The most prevalent diagnosis was CPVT as 4 genetic variants were found which were linked to a high probability of this channelopathy. In another case, SQTS was diagnosed based on a new mutation in KCNH2 with a borderline ECG (QTc = 357 ms).
In 8 cases, clinical data were obtained that guided the diagnosis, but firm diagnostic criteria were not met and the phenotype data did not completely fit with the genotype observed ( Table 3 ). The assessment of 88 relatives revealed 19 affected relatives: 10 with BS, 5 with CPVT, 3 with LQTS, and another with probable SQTS. Eighty-one were first-degree relatives, and the remaining were second- or third-degree relatives. The relatives with LQTS and CPVT were treated with β blockers. Further data on these positive relatives can be found in Supplementary Table 2 .
| Age-Sex | Cause of Ventricular Fibrillation/syncope | Previous syncope | QTc (ms) | Mutation | Relatives assessed/ positive for variant | Diagnosis suspected because of: | |
|---|---|---|---|---|---|---|---|
| Case 1 | 45-Male | No | No | 395 | LMNA (Arg454His) | 0/0 | Laminopathy: first degree AV block + distal polyneuropathy |
| Case 2 | 50-Male | Exercise | No | 410 | DSG – 2 (Ser30Arg) | 4/4 | Suspected left arrhythmogenic cardiomyopathy (2 minor criteria) |
| Case 3 | 32-Male | Stress/alarm clock | No | 432 | KCNQ1 (477 + 4C>T) RyR2 (Pro1857Leu) | 0/0 | Overlap Long QT Syndrome-Catecholaminergic Polymorphic Ventricular Tachycardia: co-existence of atrial fibrillation with amiodarone + cause of ventricular fibrillation |
| Case 4 | 43-Female | Asthma inhalers | No | 440 | MYBPC3 (Glu619Lys) (Gly690Arg) | 0/0 | Hypertrophic cardiomyopathy: double mutation, described as pathogenic No MRI |
| Case 5 | 39-Male | Post-exercise | No | 421 | SCN5A (IVS3-5 C>A) | 1/0 | Brugada syndrome: mutation previously associated with BS on 2 occasions, and LQTS on 1 |
| Case 6 | 16-Female | Unknown | No | 390 | KCNH2 (Gly21Asp) | 3/1 | Long QT Syndrome: only variant detected, molecular characteristics suggest pathogenicity |
| Case 7 | 63-Female | Unknown | No | 383 | RyR2 (Ser1765Cys) | 0/0 | Catecholaminergic Polymorphic Ventricular Tachycardia: only variant detected, molecular characteristics suggest pathogenicity |
| Case 8 | 23-Female | Unknown | No | 453 | AKAP – 9 (Arg3450Gln) | 1/1 | Long QT Syndrome: very infrequent in controls, molecular characteristics suggest pathogenicity |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


