G. Michael Felker, John R. Teerlink
Diagnosis and Management of Acute Heart Failure
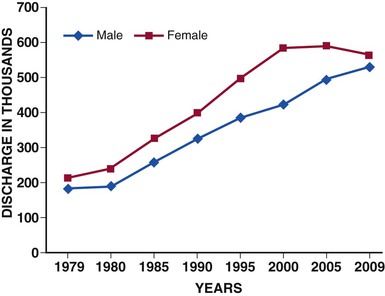
Acute heart failure (AHF) is among the most common causes for hospitalization in patients older than 65 years of age in the developed world. In the United States alone, approximately 3 million patients are hospitalized each year with a primary or secondary diagnosis of heart failure, and AHF contributes to more than 7 million hospital days annually.1 The number of hospitalizations for heart failure has tripled during the past three decades (Fig. 24-1) and is projected to continue to increase because of a convergence of several epidemiologic trends: the aging of the population, against a background of the age-related incidence of heart failure; the reduction in hypertension-related mortality and the greatly improved survival after myocardial infarction, resulting in more patients living with chronic left ventricular (LV) dysfunction; and the availability of effective therapy for prevention of sudden death. Previously considered part of the clinical history of chronic heart failure, AHF is increasingly recognized as a distinct disorder with unique epidemiology, pathophysiology, treatments, and outcomes.
Epidemiology
Nomenclature and Definition
A variety of overlapping terms have been used to characterize AHF in the literature, including “acute heart failure syndromes” (AHFSs), “acute(ly) decompensated heart failure” (ADHF), “acute decompensation of chronic heart failure” (ADCHF), and “hospitalization for heart failure” (HHF). Although none of these is universally accepted, the term acute heart failure is used in this chapter for consistency and simplicity. Broadly speaking, AHF can be defined as the new onset or recurrence of symptoms and signs of heart failure requiring urgent or emergent therapy and resulting in seeking unscheduled care or hospitalization. Although the designation “acute” in the nomenclature suggests a sudden onset of symptoms, many patients may have a more subacute course, with gradual worsening of symptoms that ultimately reach a level of severity sufficient to seek unscheduled medical care.
Scope of the Problem
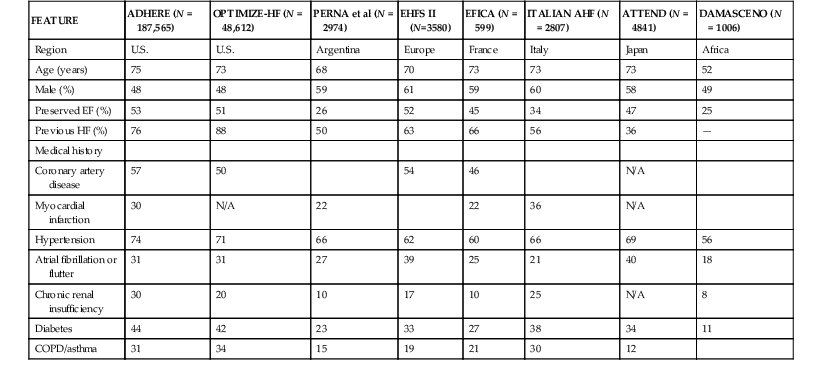
AHF represents a major burden in the developed world. In the United States, heart failure is the primary diagnosis for more than 1 million hospitalized patients annually, and a secondary diagnosis in an additional 2 million hospitalizations.1 Even greater numbers of hospitalizations are reported in Europe.2 The annual direct and indirect costs associated with heart failure approach $40 billion in the United States, and most of these expenditures are related to the costs of hospitalizations. As noted previously, the overall number of hospitalizations for heart failure continues to grow as a consequence of the aging of the population, improved survival after acute myocardial infarction, and effective prevention of sudden cardiac death. Recent data, however, suggest that the age-adjusted rate of hospitalization for heart failure may have begun to decrease with improvements in chronic heart failure therapy. In a study using U.S. Medicare claims data from 1998 to 2008, age-related incidence of hospitalization for heart failure declined for all race and gender groups.3 Similar data have been published in several countries in Europe.4 Despite these potentially encouraging trends, it appears likely that heart failure–related hospitalization will be a major clinical and economic problem for health care systems for the foreseeable future. A major breakthrough in the understanding of the epidemiology, clinical characteristics, and outcomes of patients with AHF has been the development of large, relatively unselected registries of AHF cases throughout the world, which have provided a “real world” perspective on the epidemiology and outcomes of this clinical syndrome (Table 24-1).
Preserved Versus Reduced Ejection Fraction
As with chronic heart failure, recent decades have seen increasing recognition of the epidemiologic importance of heart failure with normal or near-normal systolic function, so-called heart failure with preserved ejection fraction (HFpEF) (see also Chapter 27). On the basis of the registry data, 40% to 50% of patients hospitalized have HFpEF.5 The unexpectedly high prevalence of HFpEF in AHF registries exemplifies the way registry data can better inform understanding of the epidemiology of a clinical problem compared with clinical trials, which have tended to focus on younger patients with impaired systolic function and fewer comorbid diseases. Compared with patients with heart failure and low ejection fraction, those with HFpEF are more likely to be older, to be female, and to have a history of hypertension, while being less likely to have underlying coronary artery disease.5 Although these differences are notable in large epidemiology studies, it is important to recognize that individual patient characteristics, clinical presentation, and physical examination are not sufficient to distinguish these entities at the bedside without formal assessment of ejection fraction. The in-hospital mortality for patients with HFpEF appears to be lower than that for patients with depressed LV ejection fraction (LVEF), but postdischarge rehospitalization rates are similarly high for both groups. Patients with AHF and HFpEF are more likely to be rehospitalized for and to die from noncardiovascular causes than patients with AHF and reduced ejection fraction, reflecting their more advanced age and greater burden of comorbidity.6
Age, Race, and Sex
Significant differences in the epidemiology of AHF, based on age, race, and sex, have been recognized. AHF disproportionally affects elderly people, with a mean age of 75 years in large registries. In clinical trial populations, by contrast, the mean age often is substantially lower. AHF affects men and women almost equally, but important differences by sex are recognized. In the ADHERE registry, women admitted for AHF were older than men (74 versus 70 years), and more frequently had preserved systolic function (51% versus 28%).7 Differences in ethnic groups have been studied most extensively in the United States and have focused primarily on differences between African American and white patients. In the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) registry, African American patients admitted with AHF were younger (64 versus 75 years), more likely to have LV systolic dysfunction (57% versus 51%) with a lower mean ejection fraction (35% versus 40%), hypertensive cause for heart failure (39% versus 19%), renal dysfunction, and diabetes, compared with the non–African American group.8 Lower crude mortality rates have been reported for African Americans than for non–African American patients, but when adjustments are made for these differences in comorbidity and age, mortality rates are similar.
Comorbid Conditions
Concomitant diseases are very common in patients hospitalized with AHF, reflective of the elderly population. These comorbid conditions not only represent diseases that are risk factors for the development of heart failure but also can complicate diagnosis and management. Hypertension is the most prevalent of the concurrent conditions, present in approximately two thirds of the patients (see also Chapters 43 and 44); coronary artery disease is present in approximately half and dyslipidemia more than one third (see also Chapter 54).9,10 Other conditions that are the result of the vascular injury produced by these diseases, such as stroke, peripheral vascular disease, and chronic renal insufficiency, also are very common in patients with AHF. Diabetes mellitus is present in greater than 40% of U.S. patients, most likely related to increasing incidence of obesity, and in 27% to 38% of patients in Europe. Chronic obstructive pulmonary disease (COPD) also is present in approximately 25% to 30%, which confounds the presenting symptoms of dyspnea and is associated with lower use of evidence-based therapy.11 Atrial fibrillation appears to be more common in Europe (reported frequency of up to 42%, compared with 31% in U.S. patients with AHF), and can both precipitate AHF and complicate its management.12
Pathophysiology
AHF is not a single disease but a heterogeneous clinical syndrome. Accordingly, the pathophysiology of AHF is complex and highly variable, with many overlapping pathogenic mechanisms that may be operative in a given clinical scenario to a greater or lesser degree. This fundamental heterogeneity complicates the attempt to create a simple and unified conceptual model. One potentially useful framework for understanding of the pathophysiology of AHF is to consider it as the result of the interaction of underlying substrate, initiating mechanisms or triggers, and amplifying mechanisms, all of which contribute to a common set of clinical signs and symptoms (primarily related to congestion or end-organ dysfunction, or both) that define AHF (Fig. 24-2). In this context, substrate refers to underlying cardiac structure and function. The underlying substrate may be one of normal ventricular function, as in patients without a previous history of heart failure in whom AHF develops because of sudden changes in ventricular function from an acute insult such as myocardial infarction or acute myocarditis. Alternatively, some patients may have no previous history of heart failure but exhibit an abnormal substrate (e.g., those with stage B heart failure associated with asymptomatic LV dysfunction) with a first presentation of heart failure (de novo heart failure). Finally, in most patients with AHF, the original substrate is one of chronic compensated heart failure, followed by decompensation with development of AHF.
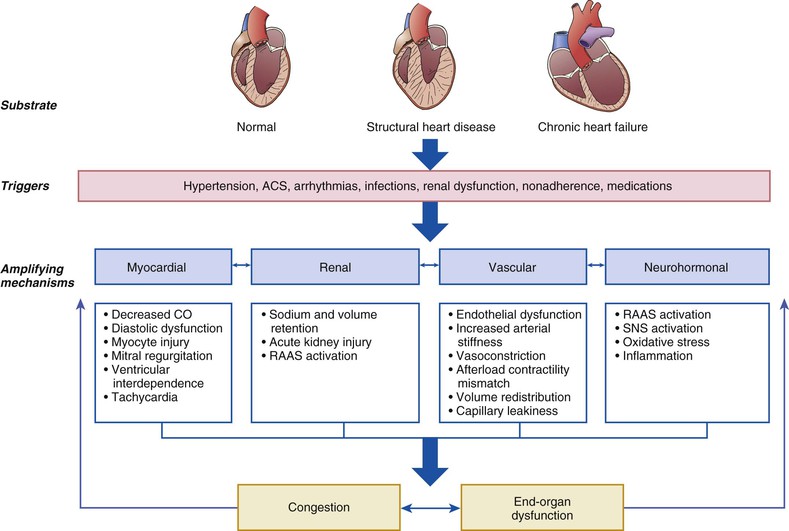
Initiating mechanisms vary according to, and interact with, the underlying substrate and may be cardiac or extracardiac. For patients with normal substrate (normal myocardium), a substantial insult to cardiac performance (e.g., acute myocarditis) is required to lead to the clinical presentation of AHF. For patients with abnormal substrate at baseline (e.g., asymptomatic LV dysfunction), smaller perturbations (e.g., poorly controlled hypertension, atrial fibrillation, or ischemia) may precipitate an AHF episode. For patients with a substrate of compensated or stable chronic heart failure, medical or dietary noncompliance, drugs such as nonsteroidal anti-inflammatory agents or thiazolidinediones, and infectious processes all are common triggers for decompensation.
Regardless of the substrate or initiating factors, a variety of “amplifying mechanisms” perpetuate and contribute to the episode of decompensation. These include neurohormonal and inflammatory activation, ongoing myocardial injury with progressive myocardial dysfunction, worsening renal function, and interactions with the peripheral vasculature, all of which may contribute to the propagation and worsening of the AHF episode.
Congestion
Systemic or pulmonary congestion, most often the result of a high LV diastolic pressure, dominates the clinical presentation in most patients hospitalized for AHF. In this sense, congestion can be seen as a final common pathway by which mechanisms described in this section produce clinical symptoms leading to hospitalization. A simplified view of AHF pathophysiology is that gradual increases in intravascular volume lead to symptoms of congestion and clinical presentation, and normalization of volume status with diuretic therapy results in restoration of homeostasis. Although this mechanism may be operative in some patients (particularly those with frank noncompliance with sodium restriction or diuretic therapy), this model is a vast oversimplification. Congestion often occurs even in the absence of nonadherence, and the same degree of nonadherence may not lead to decompensation in a given patient. Although some data suggest that increases in body weight often precede decompensation and hospitalization for heart failure, careful studies using implantable hemodynamic monitors suggest that increases in invasively measured LV filling pressures can occur without substantial changes in body weight.13 These observations have led to increasing interest in the concept of volume redistribution rather than volume retention as a mechanism of decompensation in heart failure (discussed in more detail in a later section, Vascular Mechanisms).
One potentially important concept is the distinction between clinical congestion and hemodynamic congestion. Although patients present with signs and symptoms of systemic congestion such as dyspnea, rales, elevated jugular venous pressure, and edema, this state often is preceded by so-called hemodynamic congestion, defined as high LV diastolic pressures without overt clinical signs. Similarly, clinical congestion may resolve with treatment but hemodynamic congestion may persist, leading to a high risk of rehospitalization. It has been postulated that hemodynamic congestion may contribute to the progression of heart failure because it may result in wall stress, as well as in renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS) activation. These effects may trigger a variety of molecular responses in the myocardium, including myocyte loss and increased fibrosis. Concomitant abnormal processing of the natriuretic peptides, which are the intrinsic counterregulatory hormones in heart failure, leads to diminished biologic activity in patients with advanced disease.14 In addition, elevated diastolic filling pressures may decrease coronary perfusion pressure, resulting in subendocardial ischemia with further exacerbation of cardiac dysfunction. Increased LV filling pressures also can lead to acute changes in ventricular architecture (resulting in a more spherical shape), contributing to worsening mitral regurgitation. These mechanisms also play an important role in pathologic remodeling of the ventricle, a chronic process that may be accelerated by each episode of decompensation. Consistent with this paradigm is the well-established clinical observation that each hospitalization for AHF heralds a substantial worsening of the long-term prognosis, an effect that appears additive with recurrent hospitalizations.15 Data from studies with implantable hemodynamic monitors have confirmed that chronically elevated filling pressures (i.e, hemodynamic congestion) are associated with increased risk of future events.16 Although congestion is widely recognized at the most common aspect of AHF presentation, only recently has there been a formal attempt to better assess and quantitate congestion in heart failure.17
Myocardial Function
Although a variety of extracardiac factors play important roles in AHF, impairments of cardiac function (systolic, diastolic, or both) remain central to our understanding of this disorder. Changes in systolic function and decreased arterial filling can initiate a cascade of effects that are adaptive in the short-term but maladaptive when elevated chronically, including stimulation of the sympathetic nervous system and the renin-angiotensin-aldosterone axis. Activation of these neurohormonal axes leads to vasoconstriction, sodium and water retention, increase and redistribution from other vascular beds, increases in diastolic filling pressures, and clinical symptoms. In patients with underlying ischemic heart disease, initial defects in systolic function may initiate a vicious circle of decreasing coronary perfusion, increased myocardial wall stress, and progressively worsening cardiac performance. Increased LV filling pressures and changes in LV geometry can worsen functional mitral regurgitation, further decreasing cardiac output.
Although decreases in systolic function can clearly play a role in the pathophysiology of AHF, epidemiology data summarized earlier show that approximately half of patients with AHF have relatively preserved systolic function. Of importance, abnormalities in diastolic function are present in patients with both preserved and impaired ejection fraction. The impairment of the diastolic phase may be related to passive stiffness or abnormal active relaxation of the left ventricle, or both. Hypertension, tachycardia, and myocardial ischemia (even in the absence of coronary artery disease) can further impair diastolic filling. All of these mechanisms contribute to higher LV end-diastolic pressures, which are reflected back to the pulmonary capillary circulation. Diastolic dysfunction alone may be insufficient to lead to AHF, but it serves as the substrate on which other precipitating factors (such as atrial fibrillation, coronary artery disease, or hypertension) lead to decompensation. One underappreciated aspect of myocardial function in AHF relates to the interdependence of the left and right ventricles. Because of the constraints of the pericardial space, distention of either ventricle secondary to increased filling pressures can result in direct impingement of diastolic filling of the other ventricle. This mechanism may be particularly operative in clinical scenarios leading to abrupt failure of the right ventricle (such as pulmonary embolism or right ventricular [RV] infarction), resulting in diminished filling of the left ventricle and arterial hypotension.
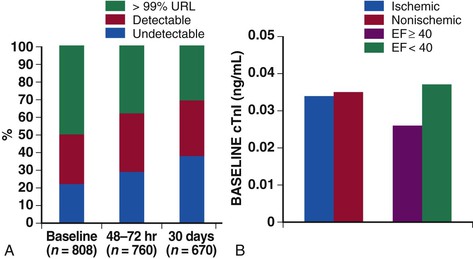
The availability of sensitive assays for circulating cardiac troponins has led to substantial evolution of our understanding of the role of myocardial injury in the pathophysiology of heart failure. Data from both registries and clinical trial populations indicate that circulating cardiac troponins are elevated in a large proportion of patients with AHF, even in the absence of clinically overt myocardial ischemia.18,19 In a representative analysis of the ASCEND-HF study, 50% of patients with AHF had troponin levels about the 99th percentile upper reference limit (the diagnostic cut point for myocardial infarction) at baseline and 30% had persistent elevation of troponin at 30 days20 (Fig. 24-3). In a smaller study, more than 20% of patients admitted with AHF and with negative troponins at baseline converted to detectable levels by day 7.21 Most studies have indicated that troponin elevations are associated with increased risk of both in-hospital and postdischarge events.
The precise mechanisms mediating myocardial injury in AHF are poorly defined, but increased myocardial wall stress, decreased coronary perfusion pressure, increased myocardial oxygen demand, endothelial dysfunction, activation of the neurohormonal and inflammatory axes, platelet activation, and altered calcium handling all may contribute to myocyte injury even in the absence of epicardial coronary artery disease.18 Specific therapeutic interventions that may increase myocardial oxygen demand (such as positive inotropic agents) or decrease coronary artery perfusion pressure (such as some vasodilators) may exacerbate myocardial injury and further contribute to the cycle of decompensation. Whether avoidance of myocardial injury is a specific target for therapy in AHF remains a subject of active investigation.
Renal Mechanisms (see also Chapter 88)
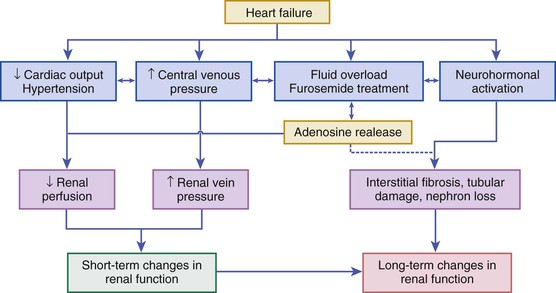
The kidney plays two fundamental roles relative to the pathophysiology of heart failure: It modulates loading conditions of the heart by controlling intravascular volume and is responsible for neurohormonal outputs (i.e., the RAAS system). Abnormalities of renal function are extremely common in patients with AHF and may be underestimated by creatinine alone—64% of patients in the ADHERE registry had a glomerular filtration rate below 60 mL/min/1.73 m2.22 Baseline measures of renal function also are well-established risk factors for poor outcomes in AHF (see Risk Stratification section later on). Additionally, worsening renal function during AHF therapy in the setting of persistent congestion—often termed the “cardiorenal syndrome”—has been associated with poor outcomes in a variety of observational studies.23 Multiple studies have investigated the pathophysiology and risk factors for this phenomenon, which is related to an intricate interplay of patient characteristics (age), comorbid disease (baseline renal function as assessed by glomerular filtration rate [GFR], diabetes mellitus, hypertension), neurohormonal activation (especially of RAAS and SNS), and hemodynamic factors (central venous congestion, and less frequently arterial underfilling with renal hypoperfusion), as well as other factors such as activation of inflammatory cascades and oxidative stress24 (Fig. 24-4). Although often assumed to be related to low cardiac output and renal blood flow, careful hemodynamic studies have confirmed that the strongest predictor of worsening renal function in heart failure patients relates to elevated central venous pressure, which is reflected back to the renal veins and leads directly to changes in glomerular filtration rate.25 Specific therapies for AHF such as diuretics may exacerbate renal dysfunction through increasing neurohormonal activation and vasoconstriction, although in many cases effective diuresis improves renal function by decreasing central venous pressure. It may be important to distinguish between changes in renal function (a potentially transient phenomenon often related to local or systemic hemodynamic factors) and frank renal injury. Transient changes in renal function often occur during AHF therapy, but these frequently are temporary and do not appear to be associated with adverse outcomes, especially in the presence of effective decongestion.26,27 Newer biomarkers that may distinguish changes in renal function (as reflected by serum creatinine or cystatin C) from acute kidney injury (as reflected by markers such as urinary neutrophil gelatinase–associated lipocalin [NGAL]) may allow better differentiation of worsening renal function during AHF hospitalization.28 A detailed classification system for understanding the interplay between cardiac performance and renal function has been proposed and provides a framework for understanding the complex pathophysiology underlying the cardiorenal syndrome.24,29 Clinical aspects of the diagnosis and management of the cardiorenal syndrome in AHF are covered in more detail later on.
Vascular Mechanisms
Although abnormalities in cardiac function traditionally have held the central position in the pathogenesis of AHF, there is increasing appreciation for the importance of the vasculature not only as an underlying cause of cardiac dysfunction (i.e., atherosclerosis, hypertension) but also as a central component of the pathogenesis of AHF. Abnormalities of endothelial function related to nitric oxide–dependent regulation of vascular tone are well described in heart failure.30 Arterial stiffness, which is related to but distinct from increased blood pressure, increases cardiac loading conditions and is associated with incident heart failure and worse outcomes. Peripheral vasoconstriction in the setting of AHF redistributes blood centrally, increasing pulmonary venous congestion and edema. As noted previously, elevated central venous pressure reduces renal function, resulting in greater fluid retention that further elevates venous pressures. Peripheral arterial vasoconstriction increases afterload, LV filling pressures, and postcapillary pulmonary venous pressures, resulting in worsening of pulmonary edema and dyspnea. This increased afterload causes greater ventricular wall stress and increased myocardial ischemia and cardiac arrhythmias. Abnormal vascular compliance also predisposes affected patients to marked blood pressure lability with relatively minor changes in intravascular volume, causing precipitous increases in afterload and ultimately in LV filling pressures, resulting in pulmonary congestion. The effects of this vascular abnormality are amplified by LV diastolic dysfunction.
The clinical observation that vasodilator treatment can ameliorate dyspnea in many acutely hypertensive patients without significant diuresis has led to the concept that afterload-contractility mismatch can lead to increased diastolic filling pressures in the setting of minimal total body volume changes. Similarly, the recognition of the large capacitance of the venous system (in particular, the splanchnic circulation) has led to increased interest in volume shifts from the “venous reservoir” into the effective circulatory volume as a potentially important and unrecognized mechanism in AHF.31,32 These shifts can be mediated by SNS activation, and this mechanism has been proposed as a potential explanation between the apparent disconnect between changes in filling pressures and changes in body weight during chronic hemodynamic monitoring.
Neurohormonal and Inflammatory Mechanisms (see also Chapter 22)
Although elevations of circulating neurohormones are well documented in patients with AHF, the precise role of neurohormonal activation in the pathophysiology of AHF remains to be fully delineated. Increased plasma concentrations of norepinephrine, plasma renin activity, aldosterone, and endothelin-1 have been reported in patients with AHF; all of these axes are associated with vasoconstriction and volume retention, which could contribute to myocardial ischemia and congestion, thereby exacerbating cardiac decompensation. Inflammatory activation and oxidative stress may also play a role. Proinflammatory cytokines such as tumor necrosis factor-alpha and interleukin-6 are elevated in patients with AHF and have direct negative inotropic effects on the myocardium as well as increasing capillary permeability and inducing endothelial dysfunction.33,34 In addition to direct effects, this activation stimulates the release of other factors, such as the potent procoagulant tissue factor and endothelin-1, which can lead to further myocardial suppression, disruption of the pulmonary alveolar-capillary barrier, and increased platelet aggregation and coagulation (potentially worsening ischemia).
Evaluation of the Patient with Acute Heart Failure
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


