(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Heart failure is common in patients with diabetes, even in the absence of coronary artery disease or hypertension. Diabetic cardiomyopathy is one of the leading causes of heart failure among diabetic patients. Metabolism of myocardium is altered in diabetes, which likely contributes to decreased contractile activity and ventricular failure. The mitochondria play a major role in cellular metabolism, and there is evidence indicating that mitochondrial dysfunction is critical in the pathogenesis of diabetic cardiomyopathy.
Up to 1 % of patients with diabetes are affected by maternally inherited diabetes caused by genetic abnormalities in mitochondrial DNA. Several mitochondrial DNA mutations not only are associated with mitochondrial diabetes but also are a cause of the developing cardiomyopathy.
In this chapter we will focus on the effects of diabetes on cardiac myocardium with specific reference to the role of modulation of cardiac mitochondrial function in the disease process. Several potential mechanisms that lead to mitochondrial dysfunction in the diabetic heart will be discussed.
Introduction
Patients with diabetes may develop cardiac dysfunction and heart failure (HF) in the absence of vascular disease (coronary artery disease, hypertension), a phenotype that can be named diabetic cardiomyopathy [1]. Although the underlying pathogenesis is incompletely understood, this may include dysregulated lipid metabolism, insulin resistance, mitochondrial dysfunction, and abnormalities in adipokine secretion and signaling. These abnormalities lead ultimately to impaired calcium homeostasis and finally to defects in myocardial relaxation and contractility.
Although the pathophysiological mechanisms of diabetes leading to the development of HF are certainly multifactorial, emerging evidence suggests that abnormalities in cardiac energy metabolism play a fundamental role in the development of diabetic cardiomyopathy. Increased myocardial fatty acid use and reduced glucose oxidation in diabetes are associated with increased myocardial oxygen consumption. Thus, cardiac efficiency is reduced in diabetic hearts.
One potential mechanism for reduced cardiac efficiency in diabetic hearts involves the increase in β-oxidation of fatty acids in mitochondria leading to the increased delivery of reducing equivalents to the electron transport chain. However, a limitation in oxidative phosphorylation components results in an increased ROS production by mitochondria, which in turn uncouples mitochondria and reduces the efficiency of ATP generation. Also, increased mitochondrial ROS target proteins and lipids within mitochondria, and their oxidative modification alters the integrity of the mitochondrial inner membrane, facilitating further dysfunction in mitochondria.
Increased ROS production is associated with increased apoptosis of cardiomyocytes by p53 and the activation of the cytochrome c-activated caspase-3 pathway. ROS-mediated cell death could promote abnormal cardiac remodeling, which may contribute to the characteristic morphological and functional abnormalities that are associated with diabetic cardiomyopathy [2].
Another consequence of elevated mitochondrial ROS generation is an activation of maladaptive signaling pathways in the myocardium via accumulation of upstream glycolytic intermediates. As a result, concentration of upstream glycolytic intermediates increases leading to decrease of NADH (increased aldose reductase pathways), pathological induction of certain transcription factors (via hexosamine pathway), activation of protein kinase C (via increased diacylglycerol), and increased formation of glucose-derived advanced glycation end products. Activation of damaging pathways may lead to cardiac cell death, which could contribute to the pathogenesis of diabetic cardiomyopathy [3].
Although diabetes mellitus type 2 is a multifactorial disorder, about 1% of cases represent a single gene disorder resulting from a single gene, a mitochondrial tRNALeu(UUR), mutated at position 3243. This disease is called mitochondrial diabetes [4]. Several recent reports indicate cardiac involvement in the mitochondrial diabetes which leads to the development of cardiomyopathy [5].
In this chapter we describe potential mechanisms that lead to mitochondrial dysfunction in the diabetic heart and discuss how these mitochondrial alterations may impact the development of cardiomyopathy.
Diabetes-Related Alterations in the Mitochondrial Metabolic Milieu
Several rodent models of type 1 and type 2 diabetes have shown myocardial dysfunction associated to metabolic changes in cardiomyocytes and myocardial mitochondria. For instance, reduced expression of ETC complexes and impairment in myocardial mitochondrial ATP synthesis rates was demonstrated in diabetic type 1 and type 2 mice [6–11]. Although limited human studies have addressed the pathogenesis of diabetic cardiomyopathy, emerging data demonstrate that dysfunction of mitochondria occurs in the hearts of humans with diabetes or insulin resistance. Thus, young females with obesity and insulin resistance, studied by Peterson et al. [12], demonstrated reduced cardiac efficiency (cardiac work required increased O2 consumption) and increased fatty acid oxidation. Since both of these processes are attributed to the mitochondria, this observation indicates alterations in cardiac mitochondrial functioning. Recently mitochondrial dysfunction was directly demonstrated in diabetic type 2 patients: in all cases impaired mitochondrial respiratory capacity of permeabilized atrial myofibers was observed, which was accompanied by an increase of mitochondrial hydrogen peroxide production, depletion of glutathione, and increased levels of hydroxynonenal- and 3-nitrotyrosine-modified proteins [13].
Under normal conditions, the tremendous energy need of the heart is compensated by fatty acids (FAs) and glucose as a preferential energy substrate. Approximately 70% and 30% of ATP is generated by the heart from fatty acids and glucose, respectively, and the level of ATP content remains constant in healthy heart. In diabetes, glucose utilization in the heart is diminished because of insulin resistance, impaired pyruvate dehydrogenase (PDH) activity, and reduced glucose transporter (Glut4) content [14]. Synthesis of ATP by diabetic cardiac mitochondria relies on mitochondrial fatty acid β-oxidation (FAO) [15, 16] which may result in impaired mitochondrial respiratory function and ultrastructure [17, 18]. The enhancement of FA oxidation is caused mainly by the increase of plasma membrane content of FA transporters in diabetic heart. In db/db heart, for instance, two FA transporters, FA translocase/CD36 (FAT/CD36) and plasma membrane fatty acid-binding protein (FABPpm), are increased in sarcolemma. Interestingly, while sarcolemmal FABPpm increases due to elevated expression of this transporter [19], level of FAT/CD36 in sarcolemma increases as a result of it translocation [19–21]. The genetic depletion of the CD36 rescues the insulin-resistant phenotype evident in mice with cardiac-restricted overexpression of PPARα, further supporting the role of FA uptake in propagating insulin resistance in the heart [22]. Therefore, redistribution of FAT/CD36 and FABPpm to cardiomyocyte plasma membrane in db/db hearts leads to increase of FA uptake and subsequent increase of FA metabolism in mitochondria.
The prevailing hypothesis for FAO-dependent deleterious functional consequences is that FAO-associated increase in electron transfer donors (NADH and FADH2) increases the electron flux through the mitochondrial electron transport chain and proton gradient across mitochondrial inner membrane (hyperpolarization). High electrochemical potential generated by the proton gradient leads to partial inhibition of the electron transport in complex III, resulting in an accumulation of reduced quinone, partial reduction of O2 to generate superoxide anion. Generation of ROS is an important source for oxidative injury and mitochondrial dysfunction in diabetes (see section below) (Fig. 20.1).
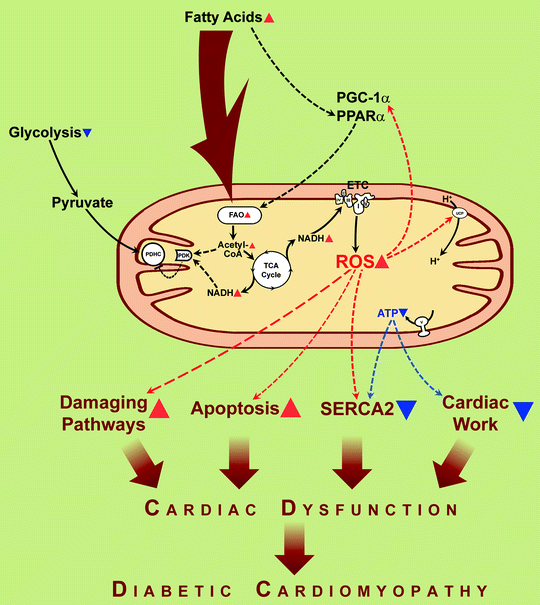
Fig. 20.1
Fatty acids induce mitochondrial uncoupling and impaired cardiac efficiency. In diabetes, increased delivery of fatty acids to the myocardium results in increased fatty acid uptake and oxidation (FAO). The resulting increase in reducing equivalent delivery (NADH is depicted) to the electron transport chain (ETC) increases reactive oxygen species (ROS) production, leading to activation of uncoupling proteins (UCPs) and proton leak. The resulting increase in mitochondrial uncoupling leads to cardiac energy deficit (ATP synthesis does not increase proportionately to increased mitochondrial O2 consumption), impairment of cardiac relaxation (reduced capacity of sarco(endo)plasmic reticulum Ca2+-ATPase to translocate Ca2+) (“SERCA2”), and contractile dysfunction (“Cardiac work”). Increased fatty acids and ROS activate signaling via transcriptional regulators, peroxisome proliferator-activated receptor α (PPARα), and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), leading to the increased transcription of many genes involved in the oxidation of fatty acids, thereby increasing FAO even further. Resulting FAO products, NADH and acetyl-coenzyme A (acetyl-CoA), activate pyruvate dehydrogenase kinase (PDK) that phosphorylates and hence inactivates pyruvate dehydrogenase complex (PDHC) leading to diminishing of the use of glucose for energy generation. Mitochondria-originated ROS play a critical role in several pathways involved in the pathogenesis of diabetic cardiomyopathy, including damaging pathways and apoptosis. Translocations of molecules and metabolic conversions are shown by solid arrows; regulatory effects are shown by dashed arrows. Red arrowheads depict increases of compounds, activation of processes. Blue arrowheads depict decreases of compounds, inactivation of processes
Also, dramatic increase of FAO leads to increase of citrate in mitochondria, inhibition of phosphofructokinase, and therefore inhibition of glycolysis [23] (Fig. 20.2). Another consequence of high level of fatty acids supply is their incorporation into triglycerides (TAG) and an accumulation of TAGs in myocardium [24–26].
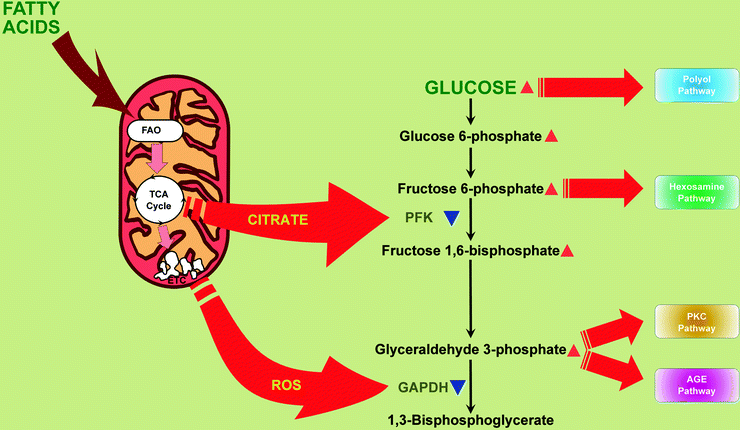
Fig. 20.2
Mitochondria contribute to hyperglycemia-induced activation of hyperglycemic damage pathways in myocardium. Increased mitochondrial citrate and reactive oxygen species (ROS) partially inhibit glycolytic enzymes, phosphofructokinase (PFK), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), respectively. Under these conditions, upstream metabolites divert from glycolysis into pathways of glucose overutilization. This results in increased glucose flux through the polyol pathway (leads to consuming of NADPH and depletion of reduced glutathione) and increased flux of fructose-6-phosphate through hexosamine pathway (leads to modification of proteins by O-linked N-acetylglucosamine). Increased triose phosphates convert into diacylglycerol (DAG), an activator of protein kinase C (PKC) isoforms (activation of PKC pathway), and to methylglyoxal, the main intracellular advanced glycation end product (AGE) precursor (activation of AGE pathway). Other abbreviations: FAO fatty acid β-oxidation, TCA cycle tricarboxylic acid cycle, ETC electron transport chain. Red arrowheads depict increases of compounds. Blue arrowheads depict inactivation of enzymes
As diabetes progresses, increased delivery of FAs activates peroxisome proliferator-activated receptor alpha (PPARα) signaling pathways, which leads to transcriptional induction of enzymes involved in FA uptake, FAO, and increased expression of PDH kinase (PDK) 4, which further suppresses glucose oxidation by decreasing PDH activity [9, 14, 15, 24, 27, 28] (Fig. 20.1). PPARα-null mice have diminished muscle fatty acid β-oxidation and resist lipid-induced diabetes [29].
There is also evidence linking Ca2+-independent isoform of phospholipase A2 (iPLA2) with cardiac injury in diabetic cardiomyopathy. This enzyme is distributed in mitochondrial membranes and catalyzes the release of fatty acids from phospholipids. A significant increase was reported in both iPLA2 mRNA and iPLA2 activity with diabetes in rat myocardium [30], thus contributing to abnormal elevation of FAO. In addition, iPLA2 mediates accumulation of signaling lipids (endocannabinoid precursors, 2-arachidonoyl lysophosphatidylcholine), influencing mitochondrial bioenergetics and fat oxidation [31, 32]. iPLA2-null mice exhibit remarkable resistance to obesity and metabolic abnormalities in obese animals associated with impaired mitochondrial fatty acid β-oxidation [33], suggesting that the enzyme plays an important role in nutritional effects on metabolic regulation.
Because fatty acids compare to glucose is an inefficient substrate, increased FAO in diabetic heart is accompanied by an increase in myocardial oxygen consumption and reduced cardiac efficiency [12, 16, 34, 35]. Recent studies of Wang et al. [36] demonstrate that acceleration of ADP/ATP transport between cardiac mitochondria and cytosol significantly improves cardiac function in streptozotocin (STZ)-treated mice: cardiomyocyte-restricted overexpression of adenine nucleotide translocator 1 (ANT1) prevents the development of diabetic type 1 cardiomyopathy probably via improvement of the cardiac cell supply with ATP on demand. According to Veksler et al. [37], functional activity of mitochondrial creatine kinase is also depressed in STZ-treated rats and may be associated with low creatine-stimulated respiration and the development of diabetic cardiomyopathy.
One of the processes in cardiomyocytes which is ATP dependent is a sequestration of cytosolic Ca2+ by SERCA2. Decrease of ATP production by hyperglycemic mitochondria limits activity of SERCA2 (Fig. 20.1), and this leads to impaired uptake of Ca2+ from cytosol to sarcoplasmic reticulum and impaired diastolic relaxation. Improvement of mitochondrial ATP production in hyperglycemic conditions via restoration of the level of cytochrome c oxidase subunit 1 (see section below) has beneficial effect in the cardiac cell energy balance and recovers cytosolic calcium handling [38].
As the regulatory control of mitochondrial bioenergetics has been identified, therapeutic agents directed at modulating mitochondrial function in the heart are being tested. For instance, PPARγ agonist (rosiglitazone) has been shown to improve myocardial efficiency in db/db mice by normalizing plasma glucose and fatty acid levels and restoring cardiac substrate utilization and insulin signaling [39, 40]. Overexpression of another isoform of PPAR, PPARδ, increases glucose uptake and oxidation [41] which potentially can ameliorate the development of insulin resistance or diabetes type 2.
Role of Mitochondria-Derived ROS in Diabetic Myocardium
An important mechanism for the pathophysiology of diabetes and its complications is increased oxidative stress (OS) [42, 43]. For instance, hyperglycemia is associated with the production of advanced glycation end products (AGEs) [44, 45] which induce production of cytosolic reactive oxygen species (ROS) via AGE receptor (RAGE). Cytosolic ROS may damage mitochondria by opening the mitochondrial permeability transition pore (MPTP) [44, 46] and by enhancement of mitochondrial ROS production [44]. Shen et al. [47] recently showed that the small GTP-binding protein Rac1 and NADPH oxidase play crucial role in the hyperglycemic elevation of cytosolic ROS with subsequent stimulation of mitochondrial ROS production. Deficiency in Rac1 or inhibition of NADPH oxidase significantly reduced hyperglycemia-induced mitochondrial ROS production in the mouse model of STZ-induced diabetes. Authors suggested that high glucose stimulates Rac1-dependent translocation of cytosolic p67phox subunit of NADPH oxidase to the sarcolemma for the assembly of active NADPH oxidase, leading to superoxide anion generation. NADPH oxidase-derived superoxide directly activates the mitoKATP channels, forms peroxynitrite, and dismutates to hydrogen peroxide—factors known to damage respiratory complexes, leading to mitochondrial ROS production. Thus, Rac1–NADPH oxidase–mitochondrion axis is important in ROS production and cardiomyocyte apoptosis in hyperglycemia, which may contribute to the development of diabetic cardiomyopathy.
Interestingly, increased mitochondrial ROS generation has been described in hearts of db/db mice (diabetes type 2 animal model) but not in Akita mouse model of diabetes type 1 [6, 48]. An important difference between the hearts of db/db mice vs. Akita mice is an insulin resistance in obese model, whereas in diabetes type 1 model, insulin sensitivity is preserved. This raises the intriguing possibility that myocardial insulin resistance may specifically prompt cardiac mitochondria to ROS overproduction via mechanisms that have to be clarified. In agreement with this hypothesis are the observations of Boudina et al. [49] that cardiac-specific deletion of insulin receptor leads to increase of hydrogen peroxide production. Recent studies on Akita mouse model of diabetes type 1 revealed the role of the growth factor adapter protein p66ShcA in the hyperglycemia-induced damage to cardiac cells sensitive to insulin. According to Malhotra et al. [50], hyperglycemia induces phosphorylation of serine residue of p66ShcA at critical position 36 (phospho-Ser36), facilitating its translocation to mitochondria where p66ShcA generates ROS via the oxidation of cytochrome c. Cytochrome c-originated ROS cause the opening of the MPTP, a collapse of mitochondrial transmembrane potential, and activation of the intrinsic apoptosis program (see below). Interventions that interrupt or prevent phosphorylation at Ser36 of the p66ShcA protein improve the resistency of cells to an oxidant stress, stabilize mitochondrial energetics, and prevent hyperglycemia-caused apoptosis.
The significance of mitochondria generated ROS in diabetes has been proposed by several laboratories [51–55]. ROS generated by the mitochondria can directly damage proteins and mitochondrial DNA by oxidation and oxidize lipids to form lipid peroxidation products, which can induce protein or phospholipid damage. In several studies, the role of mitochondrial oxidative stress has been confirmed in diabetic cardiomyopathy (Fig. 20.1). Thus, overexpression of antioxidant protein (metallothionein) or superoxide scavengers (catalase, mitochondrial manganese superoxide dismutase (MnSOD)) reduces nitrosative damage, preserves mitochondrial morphology, and rescues cardiac contractile dysfunction in animal models of diabetes types 1 and 2 [56–59]. The fact of interorganelle antioxidant protection (cytoplasmic antioxidants, catalase and metallothionein, protect the mitochondria; mitochondrial antioxidant MnSOD protects cytosolic structures) implies that diffusible ROS, like hydrogen peroxide, are important in diabetic cardiomyopathy. ROS also generate peroxynitrite from nitric oxide (NO), causing intracellular nitrosylation. Reactive nitrogen species (RNS) produced by peroxynitrite can nitrate tyrosines in proteins and thus change the catalytic activity of enzymes, alter cytoskeletal proteins, and impair cell signal transduction. For example, an increased tyrosine nitration of cardiac mitochondrial proteins and association of tyrosine nitration with cardiomyocyte death were demonstrated in mice with type 1 diabetes [56, 60].
One of the effects of elevated ROS is the mitochondrial uncoupling [10, 48, 49]. In ob/ob mice palmitate increased an oxygen consumption without a concomitant increase in contractile performance. Since myocardial O2 consumption increases, despite reduced expression of ETC complexes, it might indicate that FA-induced oxygen consumption in ob/ob hearts may be the consequence of mitochondrial uncoupling [10]. The mitochondrial uncoupling further increases oxygen consumption without a simultaneous increase in ATP production, thus contributing to decreased efficiency of mitochondrial ATP production and therefore contractility of cardiac cell. The mechanism of uncoupling includes ROS- or lipid peroxides-dependent activation of uncoupling proteins in cardiac mitochondria [61] (Fig. 20.1).
Also, in diabetic ZDF rats, increased ROS can alter gene expression: it contributes to the switch in expression of cardiac myosin heavy from α- to β-isoform through the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) (antioxidant treatment prevented this change) [62].
Cardiac mitochondria are represented by two spatially, functionally, and morphologically distinct subpopulations. Subsarcolemmal mitochondria are located beneath the sarcolemma, whereas interfibrillar mitochondria are embedded between myofibrils. Mitochondrial subpopulations respond differently to a diabetic stress: diabetic interfibrillar mitochondria demonstrate increased superoxide production, increased oxidative damage to proteins and lipids, decreased cardiolipin, and decreased activities of respiratory complexes. At the same time, subsarcolemmal mitochondria are practically unchanged under diabetic stress [63]. Because interfibrillar mitochondria provide ATP for the contractility apparatus, the decrease in their OXPHOS function may be detrimental to cardiac contractility, thus contributing to the contractile dysfunction associated with diabetic cardiomyopathy. Cardiac mitochondrial subpopulations also differ in susceptibility to diabetes-related apoptosis (see next section).
Thus, OS-induced damage of mitochondria and ROS overproduction in the diabetic myocardium may contribute to myocardial dysfunction.
Apoptosis
In the hearts of diabetic patients and animal models of diabetes, increased levels of apoptosis have been detected, and the development of diabetic cardiomyopathy is associated with the loss of cardiomyocytes [64, 65]. It seems that cardiomyocytes in the diabetic myocardium are more susceptible to apoptosis [64]. Loss of cardiac cells decreases the ability of the heart to maintain contractility. Under these conditions, the remaining myocytes work harder to compensate for the loss in contractility units, and this, in turn, contributes to the development of cardiac hypertrophy leading to the cardiomyopathy and HF.
Hyperglycemia- and diabetes mellitus-associated increase in reactive oxygen and nitrogen species production play a causal role in the apoptotic cardiomyocyte death, diastolic dysfunction, and the progression to HF [66]. Studies indicate that inhibition of apoptotic signaling pathways or antioxidant-dependent decrease of cardiomyocyte death significantly prevents the diabetic cardiotoxicity, which suggests that oxidative stress-induced apoptosis significantly contributes to the development of diabetic cardiomyopathy [57, 67] (Fig. 20.1).
Hyperglycemia-induced myocardial apoptosis in diabetic mice produced by STZ and H9c2 cardiac myoblast cells exposed to high levels of glucose is mediated, at least in part, by mitochondria-mediated proapoptotic events (mitochondrial cytochrome c release and activation of caspase-3), which may be triggered by ROS derived from high levels of glucose [64]. Mitochondrial derived ROS are implicated in hyperglycemia-induced Rac1/NADPH-mediated cell apoptosis in the hearts of db/db and STZ diabetic mice [47]. Similarly, phospho-p66ShcA/cytochrome c-generated ROS in hyperglycemic Akito mice cardiomyocytes (see previous section) initiate intrinsic apoptosis: phosphorylation/activation of proapoptotic transcription factor p53, expression of p53-dependent Bax, release of cytochrome c, and cleavage of pro-caspase-3 [50].
Antioxidant α-lipoic acid (α-LA) effectively attenuates mitochondria-dependent cardiac apoptosis and employs a protective role against the development of diabetic cardiomyopathy. The ability of α-LA to suppress oxidation-caused damage of mitochondria is related to an increase in the reduced glutathione content and an enhancement of manganese superoxide dismutase activity in cardiac mitochondria [68].
In isolated cardiac cells, mitochondrial fission contributes to high-glucose-induced apoptosis: inhibition of mitochondrial fission (overexpression of a dominant-negative dynamin-like protein) normalizes mitochondrial morphology, ROS levels, and cell death [69].
One potential mechanism of cardiac cell death associated with diabetes type 2 and obesity, and thus contributing to cardiac dysfunction, includes alterations of mitochondrial membrane phospholipid composition by increased uptake of long-chain FAs. For instance, palmitate induces apoptosis in rat cardiomyocytes by diminishing concentration of cardiolipin in mitochondria [70] and via de novo ceramide formation/mitochondrial apoptotic pathway activation [71]. Change of the dynamics of mitochondrial membranes modified by FAs may lead to the detachment of cytochrome c from the mitochondrial inner membrane and release from mitochondria which is a necessary step for initiation of apoptosis.
In STZ-treated mice (diabetes type 1) increase of caspase-3 and caspase-9 activities indicates enhanced apoptotic signaling in the heart. Interestingly, distinct cardiac mitochondrial subpopulations demonstrate differential apoptotic susceptibility: increase of MPTP opening, decrease of mitochondrial membrane potential, lower cytochrome c and BcL-2 levels, and increased Bax levels are associated with diabetic interfibrillar mitochondria, whereas diabetic subsarcolemmal mitochondria are not susceptible to apoptosis [72]. These differences are probably based on different resistance of mitochondrial subpopulations to diabetes-induced oxidative damages (see section above).
Mitochondria and Activation of Metabolic Damaging Pathways
Hyperglycemia-induced cell damage is a consequence of increased flux through metabolic pathways such as the polyol pathway, hexosamine pathway, advanced glycation end product (AGE) formation, and activation of protein kinase C (PKC) isoforms [3, 52, 73, 74]. All of them become activated as a result of ROS-dependent inhibition of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH): glycolytic metabolites upstream to GAPDH divert from glycolysis into pathways of glucose overutilization (Fig. 20.2). The linking factor between ROS overproduction and the inhibition of GAPDH is activation of the nuclear DNA repair enzyme poly(ADP-ribose) polymerase (PARP): increased mitochondrial ROS, via inducing nuclear DNA strand breaks, activate PARP [75, 76]. Once activated, PARP catalyzes polymerization of ADP-ribose, which accumulates on GAPDH and decreases its activity [75].
As a result of GAPDH inactivation, there is an increase of glucose flux through the polyol pathway which depletes reduced glutathione and leads to formation and accumulation of AGEs. A growing body of evidence has implicated AGEs and their receptor (RAGE) as a contributory factor in the pathogenesis of diabetic vascular complications. AGE formation in the diabetic myocardium appears to be enhanced by an activation of the first and key enzyme of the polyol pathway, aldose reductase (AR) [77]. In this pathway (an alternate route of glucose metabolism), AR mediates the NADPH-dependent reduction of the aldehyde form of glucose to sorbitol which is in turn converted to fructose by NAD+-dependent sorbitol dehydrogenase. The fructose produced by the enhanced flux of glucose thru the AR-mediated polyol pathway promotes nonenzymatic glycation of proteins [3, 78].
The critical involvement of AR in both cardiovascular and ventricular damage in diabetes has been further supported by numerous studies showing that many of the deleterious effects of diabetes on vascular and cardiac function can be reversed by treatment with AR inhibitors. Iwata et al. [79] recently suggested that posttranslational modification of the AR protein particularly at a redox-sensitive cysteine residue (Cys-298) previously shown to modulate AR enzyme activity [80] may underlie the AR activation in diabetes, although this hypothesis has not yet been proven and the mechanism by which AR activation is stimulated during diabetes/hyperglycemia remains unclear.
AGEs can mediate their effects via receptors (RAGE), activating diverse signal transduction cascades and downstream pathways, including the generation of ROS and oxidative stress [81], resulting in cytokine production, subsequently inflammatory responses evoked in various types of cells, leading to the development and progression of diabetic micro- and macroangiopathy [82, 83]. This pathway has also been suggested to play a key role in the increased sensitivity of diabetic myocardium to ischemic episodes, HF [84, 85], and the so-called diabetic cardiomyopathy [86]. Diabetes-associated AGEs have been also implicated in modification of cardiac ryanodine receptors/calcium-release channels [87]. AGE-dependent modification results in dysfunction of the type 2 ryanodine receptor calcium-release channel, reduced cardiac contractility, and may be linked to the predominant left ventricular diastolic dysfunction and cardiac relaxation abnormalities associated with type 2 diabetes [88].
Similar to glucose, increased flux of fructose-6-phosphate to UDP-N-acetylglucosamine increases O-linked β-N-acetylglucosamine glycosylation of proteins (O-GlcNAcylation); increased flux of triose phosphates elevates methylglyoxal, the main intracellular AGE precursor, and diacylglycerol, an activator of PKC [3] (Fig. 20.2). Normalizing levels of mitochondrial ROS prevents glucose-induced activation of PKC, formation of AGEs, and products of polyol pathway [52].
Hexosamine pathway abnormally activated by hyperglycemic conditions is implicated in the decrease of mitochondrial transcription factor A (mtTFA) activity and/or expression, with subsequent impairment of mitochondrial function [38, 89–92]. As a result of decreased activity of mtTFA, transcription of mitochondrial DNA-encoded genes (cytochrome b, ATP synthase subunit 6, cytochrome c oxidase subunit 1) is impaired [38, 90]. For instance, hyperglycemia reduces an expression of mtTFA-regulated cytochrome c oxidase subunit 1 (CytOx1) probably due to decreased activity of CytOx1 promoter by mtTFA. The mechanism by hexosamine pathway impairs mtTFA involves O-GlcNAcylation of mtTFA. O-GlcNAcylated mtTFA demonstrates decreased binding to promoters of regulated genes, and transcription of mitochondrial DNA-encoded genes is impaired. For instance, reduced expression of mtTFA-regulated CytOx1 under hyperglycemic conditions results from decreased binding/activation of CytOx1 promoter by O-GlcNAcylated mtTFA. Overexpression of mtTFA dramatically reduces its glycosylation, restores protein levels of CytOx1, and improves ATP production in mitochondria of hyperglycemic rat cardiomyocytes [38].
Diabetes and Cardiac Mitochondrial Calcium Handling
Animal models of type 1 and type 2 diabetes—db/db mice and STZ-induced diabetic rats—demonstrate HF secondary to acute myocardial infarction [93, 94]. There is limited literature demonstrating cardiac mitochondrial calcium mishandling in diabetic animals [95–97]. In cardiomyocytes from db/db mice, mitochondrial calcium uptake was impaired [97], whereas in STZ-induced diabetic rats mitochondrial Ca2+ was reduced because of leakage through opened MPTP (cyclosporin restored Ca2+ accumulation in diabetic hearts) [98]. Taking into account that Ca2+ uptake into the mitochondrion is important for activation of the tricarboxylic acid (TCA) cycle, for ATP production, and for local modulation of cytosolic Ca2+, it is possible that altered calcium handling by mitochondria compromises energy metabolism, thereby leading to reduced contractility, and thus contributes to diabetic cardiomyopathy.
Maternally Inherited Diabetes and Deafness
In human, the mitochondrial tRNA Leu (UUR) gene mutation (A to G transition at the base pair 3243) results in diabetes called maternally inherited diabetes and deafness (MIDD). The main process by which this mutation causes diabetes is a decrease in glucose-induced insulin secretion by pancreatic β-cells causing a premature aging of these cells. Patients with MIDD subsequently develop congestive HF due to cardiomyopathy [5, 99–102]. Studies indicate that the most probable reason for the HF in this case is mitochondrial disease since it develops in the absence of hypertension or other congenital heart disease (CHD). Cardiac dysfunction in MIDD is heterogenous: it develops with [100–102, 104, 105] or without [5, 99, 103] left ventricular hypertrophy. There are a number of other mitochondrial DNA point mutations that have been associated with diabetes and myocardial disorders (see Table 20.1), but “A to G substitution at position 3243” is the most frequent mutation of mitochondrial tRNA Leu gene causing MIDD and being often misdiagnosed as either type 1 or type 2 diabetes.
Table 20.1
Mitochondrial point mutations associated with diabetes mellitus and cardiac dysfunctions
Mutation | Type of mutation | Phenotype: diabetes | Phenotype: cardiac dysfunction | Reference |
|---|---|---|---|---|
Leucine tRNA (Leu, UUR) | ||||
A3243G | Heteroplasmic | Diabetes | Left ventricular hypertrophy, diastolic dysfunction HF | |
A3252G | Heteroplasmic | Diabetes | [111] | |
C3254G | Heteroplasmic | Diabetes | Mild cardiomegaly, dilated left ventricle | [112] |
C3256T | Heteroplasmic | Diabetes | Cardiomyopathy | |
A3260G | Heteroplasmic | ? | Enlarged left ventricle, HF | [114] |
T3264G | Heteroplasmic | Diabetes | [115] | |
T3271C | Heteroplasmic | ? | Hypertrophic cardiomyopathy | [116] |
C3303T | Homoplasmic, heteroplasmic | ? | Cardiomegaly, congestive HF | [117] |
Lysine tRNA (Lys) | ||||
A8296G | Heteroplasmic | Diabetes | [118] | |
A8344G | Heteroplasmic | Diabetes | [119] | |
T8356C | Heteroplasmic | Diabetes | [120] | |
Arginine tRNA (Arg) | ||||
A10438G | Heteroplasmic | Diabetes | [121] | |
Serine tRNA (Serine) | ||||
C12258A | Heteroplasmic | Diabetes | [122] | |
Glutamic acid tRNA (Glu) | ||||
A14693G | Homoplasmic | Diabetes | [123] | |
T14709C | Heteroplasmic | Diabetes | ||
Isoleucine tRNA (Ile) | ||||
G4284A | Heteroplasmic | Diabetes | Progressive dilating cardiomyopathy, HF | [126] |
A4300G | Homoplasmic, heteroplasmic | No diabetes | Hypertrophic cardiomyopathy | |
16S ribosomal RNA | ||||
A3156G | Homoplasmic | Diabetes | [108] | |
NADH dehydrogenase, subunit 1 | ||||
G3316A | Homoplasmic | Diabetes | [129] | |
G3357A | Homoplasmic | Diabetes | [108] | |
C3375A | Homoplasmic | Diabetes | [108] | |
T3394C | Homoplasmic | Diabetes | ||
T3398C | Heteroplasmic | Diabetes | [131] | |
NADH dehydrogenase, subunit 6 | ||||
T14577C | Heteroplasmic | Diabetes | [132] < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |