Design, Execution, and Appraisal of Clinical Trials
Brian W. McCrindle
What is Evidence-Based Medicine and why are Clinical Trials so Relevant to that Practice?
Definition and Practice of Evidence-Based Medicine
The practice of evidence-based medicine occurs when clinical decision making involves the incorporation of the best available research evidence together with the clinician’s expertise and consideration of local or individual circumstances and the patient’s preferences regarding therapies and outcomes. An additional step involves tracking, appraisal, and updating of the process. As a result, the practice is not based solely on research evidence, and should not be viewed as a directive or “cook book” approach. The sequence of steps in the practice of evidence-based clinical decision making is outlined in Table 81.1.
Both Absence and Proliferation of Evidence
The important necessary first step includes a search for applicable research evidence to inform the clinical question at hand and a critical appraisal of that evidence and subsequent synthesis. Given the exponential expansion of both available evidence (i.e., the medical literature and proliferation of journals) and the ease of means to access it (i.e., searchable databases like PubMed, proliferation of open access journals), clinicians in search of answers are often faced with having to sift through hundreds or thousands of individual articles. The task often seems very daunting, and so one may resort to expert opinion or review articles (often just another form of expert opinion) or to the claims of industry. This is actually largely what many patients are doing, making their own decisions on the basis of what they are exposed to in the media or what they are told by other laypeople. The popular “news” media are fraught with errors in both reporting the facts and their interpretation, are prone to sensationalism, and in the obligation to provide balanced viewpoints often resort to inclusion of dissenting unsupported opinions from the fringes of reason. An unregulated claim extolling the curative wonders of an alternative or complementary health product of unproven effectiveness or safety may carry more weight than the best evidence-based recommendation from their healthcare provider. Together with rising healthcare costs and stories regarding conflicts of interest and sometimes conflicting study results, many patients have become suspicious of the healthcare system and abandon therapies that are of proven benefit based on best evidence, sometimes to the detriment of their own health or that of others.
TABLE 81.1 Steps in the Practice of Evidence-Based Clinical Decision Making | |
|---|---|
|
Evidence and Clinical Trials
In order to provide the best possible care to their patients and to expertly and convincingly counsel them about that care, clinicians need to be expert users and appraisers of research evidence. One needs to be able to efficiently sift through the evidence and then be able to appraise the quality of that evidence before deciding how much weight to give it in
terms of clinical decision making. One is looking to find evidence that is applicable to the clinical scenario and answerable question at hand, but of sufficient quality such that one can be confident that the findings are valid and reliable or as close to the truth as one can get. Clinical trials, by the nature of their design, execution, and systematic approach to appraisal, provide the greatest potential for freedom from biases that result in deviations from the truth and, thus, represent or contribute to the highest levels of evidence. This is why clinical trials have a greater impact within systems for developing and reporting clinical practice guidelines or recommendations. Clinical trials are assigned the highest grades in terms of quality of evidence and have the greatest influence in informing the strength of recommendations, as noted in Table 81.2 (1).
terms of clinical decision making. One is looking to find evidence that is applicable to the clinical scenario and answerable question at hand, but of sufficient quality such that one can be confident that the findings are valid and reliable or as close to the truth as one can get. Clinical trials, by the nature of their design, execution, and systematic approach to appraisal, provide the greatest potential for freedom from biases that result in deviations from the truth and, thus, represent or contribute to the highest levels of evidence. This is why clinical trials have a greater impact within systems for developing and reporting clinical practice guidelines or recommendations. Clinical trials are assigned the highest grades in terms of quality of evidence and have the greatest influence in informing the strength of recommendations, as noted in Table 81.2 (1).
TABLE 81.2 System for Grading the Quality of Evidence and the Strength of Recommendations | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
How do Clinical Trials Achieve a High Grade of Evidence?
Well-designed and well-executed randomized clinical trials achieve a high grade of evidence for several reasons, as noted in Table 81.3.
Causality
Clinical trials provide the best evidence that any differences in the comparison of outcomes being assessed are a direct result of differences in the interventions being compared (or comparison to
nonintervention or standard of care). Clinical trials provide strong evidence for defining causality and can determine the efficacy (how well it works in a research or controlled setting), effectiveness (how well it works in clinical practice), and safety of an intervention. There are several criteria for defining causality, as noted in Table 81.4. Many of these criteria are explicitly satisfied by clinical trials, and hence clinical trials provide the best evidence that an intervention either directly or indirectly (through reduction or elimination of intermediate causal factors) is the cause of observed differences in outcomes. The design and execution of clinical trials attempt to minimize confounding factors that may influence the outcomes of interest, with the observed effects then being confidently attributable to the interventions being compared. This is achieved by random allocation of study subjects, blinding of study assignment as far as possible, standardization of interventions and tracking of any cointerventions or crossovers, continuous accounting for all study subjects, and standardization of all assessments and interpretations of outcomes. Bias can be further detected and minimized during the data analysis, with statistical adjustment for any unbalances in potentially confounding factors.
nonintervention or standard of care). Clinical trials provide strong evidence for defining causality and can determine the efficacy (how well it works in a research or controlled setting), effectiveness (how well it works in clinical practice), and safety of an intervention. There are several criteria for defining causality, as noted in Table 81.4. Many of these criteria are explicitly satisfied by clinical trials, and hence clinical trials provide the best evidence that an intervention either directly or indirectly (through reduction or elimination of intermediate causal factors) is the cause of observed differences in outcomes. The design and execution of clinical trials attempt to minimize confounding factors that may influence the outcomes of interest, with the observed effects then being confidently attributable to the interventions being compared. This is achieved by random allocation of study subjects, blinding of study assignment as far as possible, standardization of interventions and tracking of any cointerventions or crossovers, continuous accounting for all study subjects, and standardization of all assessments and interpretations of outcomes. Bias can be further detected and minimized during the data analysis, with statistical adjustment for any unbalances in potentially confounding factors.
TABLE 81.3 Advantages of Randomized Clinical Trials | |
|---|---|
|
TABLE 81.4 Criteria for Defining Causality in Relationships between Interventions and Outcomes | |
|---|---|
|
Randomization
The process of randomization, or random assignment of subjects to the study interventions, is a key feature of clinical trials that minimizes bias. In order for the intervention groups to be compared fairly, they must be as similar as possible regarding all characteristics and management other than the intervention to which they were assigned. In clinical practice, patients do not receive interventions at random. Instead, decisions are usually variably based on three aspects (best research evidence, clinical expertise, patient preferences) defining evidence-based decision making. When examining the data derived from such clinical practice in a case-series review, those patients who receive one intervention versus another will likely differ regarding some characteristics that will influence the outcomes, rendering the direct comparison of outcomes unfair due to selection bias. This differential influence on outcome can be directly causal, or may have a mediating relation between the intervention and outcome, such as confounding or interaction. Random assignment of subjects helps to minimize bias from confounding by producing groups of subjects whereby baseline characteristics are similarly randomly present between groups, not only those that were measured, but also those that were not measured. Any subsequent differences between groups after random assignment should be due to chance or random error. The degree to which this achieves equivalency of the groups depends on how many subjects are being randomized, with the larger the number of subjects randomized, the smaller the likelihood that a random unbalance will be present. The success of random assignment can be tested at the end of recruitment by comparing measured baseline characteristics between groups and identifying potentially influential differences.
Prospective and Concurrent Data Collection
All clinical trials are prospective by design, whereby the main exposure, the intervention, is applied and then the subjects are followed forward in time with assessments for the outcomes. Also, all clinical trials rely on data collection that occurs in the present or concurrent data collection as the subjects progress through the study period. This prospective and concurrent data collection allows a higher quality of data to be acquired, versus the use of secondary data such as the clinical record that characterizes the majority of observational studies. Standard definitions and measurement procedures can be applied. Central measurement and interpretation can occur in core laboratories. For more subjective measurements or outcomes, expert and independent adjudication panels can be used. Blinding is important, whereby the subjects, the study personnel and investigators, and those analyzing the data have no knowledge of the randomized assignment of the subject and which intervention was received. Blinding minimizes biases from confounding due to cointerventions. Cointerventions might occur in the application of the intervention, implementation of the protocol, application and interpretation of the measurements, and the attribution of causal relationships to adverse events. Well-meaning study personnel and investigators may provide different subjective interpretations of assessments if they know the intervention the subject was assigned. Subjects have been known to differentially report adverse events while on inert placebo if they believed that they were receiving the active study drug. Thus, clinical trials, through concurrent data collection and blinding, allow a high degree of quality control over the data collection and further opportunities to minimize random and systematic errors.
Appraisal and Regulation
The methodology of clinical trials is fairly prescribed and regimented; errors in the design, execution, or analysis are often readily detectable. If the methods and results are described in sufficient
detail, the appraisal of the validity, reliability, quality, and applicability of the findings is possible. Appraisal can inform assignment of a quality and strength grade to the evidence provided by a clinical trial. Standardized reporting of clinical trials has been adopted and required by most prominent scientific journals, which facilitate appraisal. The CONSORT (Consolidated Standards of Reporting Trials; www.consort-statement.org) Statement to which many journals adhere ensures that those aspects that inform critical appraisal are clearly described during reporting (2,3). These guidelines are also now being used by investigators in the planning of clinical trials, and some funding sources require protocols to be submitted in a CONSORT format. Since clinical trials have a more influential contribution to recommendations and evidence-based clinical care, they are subject to more regulation. Institutional ethics boards may have more explicit criteria by which they assess and approve clinical trials. Regulatory agencies may also be involved, such as the US Food and Drug Administration.
detail, the appraisal of the validity, reliability, quality, and applicability of the findings is possible. Appraisal can inform assignment of a quality and strength grade to the evidence provided by a clinical trial. Standardized reporting of clinical trials has been adopted and required by most prominent scientific journals, which facilitate appraisal. The CONSORT (Consolidated Standards of Reporting Trials; www.consort-statement.org) Statement to which many journals adhere ensures that those aspects that inform critical appraisal are clearly described during reporting (2,3). These guidelines are also now being used by investigators in the planning of clinical trials, and some funding sources require protocols to be submitted in a CONSORT format. Since clinical trials have a more influential contribution to recommendations and evidence-based clinical care, they are subject to more regulation. Institutional ethics boards may have more explicit criteria by which they assess and approve clinical trials. Regulatory agencies may also be involved, such as the US Food and Drug Administration.
Why are there not more Clinical Trials?
While clinical trials provide the best evidence, there are challenges that preclude many trials from being performed.
Resource Requirements and Conflicts of Interest
The execution of clinical trials often requires a lot of resources, and almost invariably cannot be performed without some external funding. Often, this funding may come, wholly or in part, from industry. The relationship with industry brings up issues of real and perceived conflicts of interest, when the industry sponsor or partners have a financial interest in the results of the trial. This may also be true of nonindustry funding sources.
Equipoise and Execution
The logistics of designing and executing a clinical trial are often greater than those of performing an observational study. Equipoise must exist at the beginning and throughout the study period, meaning that the investigators must be convinced that there is insufficient evidence regarding the superiority or equivalency of one intervention over another or standard of care; otherwise, it would be unethical to withhold the superior intervention from subjects. Investigators must agree to follow a common protocol, that measurements must be performed to a given standard, and that every effort is made to ensure that data collection is complete and subjects are not lost to follow-up. Often additional quality control measures are necessary, including data coordinating centers, data coordinators, core laboratories, and data and safety monitoring boards (DSMBs).
Feasibility
A major factor that keeps many important clinical trials from being performed is lack of feasibility. This is most often because there are insufficient participating subjects. This may be as a result of the clinical condition being rare with few available potential subjects, necessary but restrictive inclusion and exclusion criteria limiting the number of eligible subjects, poor recruitment or consent rates, or the outcomes being tracked are rare or take exceedingly long to become manifest. Another feature about clinical trials that informs feasibility is that one can only study a limited number of interventions, usually only two or three, within any given single trial.
Failure of Assumptions
Clinical trials are risky endeavors. One usually begins with a hypothesis, around which assumptions are made based on available preliminary trials, observational studies, extrapolations, or educated guesses. These assumptions inform how much of a differential effect on a primary outcome attributable to the interventions being studied might be expected or clinically sufficient, and how much variation or error there might be around that effect. Often, well-designed clinical trials fail because these assumptions were incorrect. When a clinical trial shows an effect that is less than anticipated from the hypothesis and often not statistically significant but still potentially of clinical relevance, or the variation around the effect is such that one cannot be confident that one has excluded a clinically relevant effect, then the results of the trial fall into a “no man’s land,” being neither definitively positive or definitively negative.
Publication Bias
The results of negative and inconclusive clinical trials are less likely to be published and contribute to the body of evidence. The absence of these trials from the published evidence leads to an unbalance toward trials with positive results, the so-called “publication bias.” The current requirement for clinical trials to be registered with a cataloging service within the public domain before starting recruitment is an attempt by journal editors to prevent this bias, as well as to make participation in clinical trials more widely available to potential subjects (4).
TABLE 81.5 Specific Challenges for Performing Clinical Trials in Pediatric and Congenital Cardiology and Cardiovascular Surgery | |
|---|---|
|
Specialty-Specific Challenges
Many challenges exist regarding clinical trials specific to pediatric and congenital cardiology and cardiovascular surgery, as outlined in Table 81.5. One of the major advances in overcoming some
of these obstacles was the funding and formation in 2001 of the Pediatric Heart Network by the US National Institutes of Health, National Heart, Lung, and Blood Institute (5). The Pediatric Heart Network is a consortium of leading North American pediatric cardiology programs, together with a data coordinating center, aimed at performing multi-institutional studies, and has successfully completed some landmark clinical trials (6,7,8,9).
of these obstacles was the funding and formation in 2001 of the Pediatric Heart Network by the US National Institutes of Health, National Heart, Lung, and Blood Institute (5). The Pediatric Heart Network is a consortium of leading North American pediatric cardiology programs, together with a data coordinating center, aimed at performing multi-institutional studies, and has successfully completed some landmark clinical trials (6,7,8,9).
Design Issues for Clinical Trials
Both the design and execution of a clinical trial can have a strong influence on the degree to which the findings from the completed study represent the truth in terms of the answer to the research question (Fig. 81.1). Errors in the design and execution may compromise the degree to which the findings have validity, reliability, and generalizability. Errors in the execution can influence the degree to which the findings from the study as it was completed are reflective of the findings if the study had been performed exactly as planned. The degree to which the actual findings can be inferred to reflect the truth in the designed study reflects internal validity. Errors in the design can influence the degree to which the findings can be inferred to reflect the actual truth or the answer to the research question, which is referred to as external validity. The path to achieving both internal and external validity, hence the truth, should be considered not only for each individual study, but also considered in light of a series of research phases, gradually building up preliminary data and evidence.
TABLE 81.6 Phases of Clinical Trials | ||||
|---|---|---|---|---|
|
Phases of Clinical Trials
The study of interventions, particularly new interventions, is usually based on a series of progressively more rigorous study designs that move from concerns related to refining the intervention and determining feasibility, to more definitive determinations of efficacy, effectiveness, and safety. Each phase provides preliminary data and evidence to inform the next phase, creating a body of evidence that will eventually inform recommendations and evidence-based clinical decision making. These phases are particularly applicable to interventions involving investigational new drugs and devices, and proceed in a defined sequence, as noted in Table 81.6. The aims of each phase are different, and inform the choice of approach and study design. Given that large-scale efficacy and effectiveness trials are risky endeavors, these phases help to ensure that sufficient rationale and preliminary data inform their design and execution.
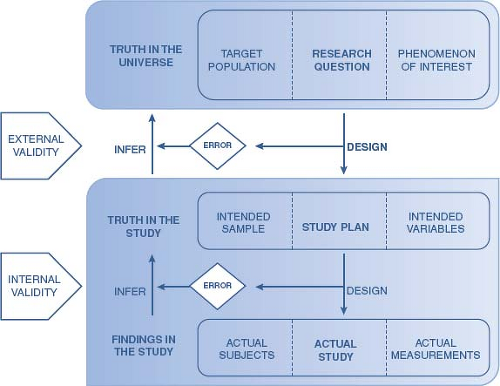 Figure 81.1 Validity and the relationship to truth in clinical trials. |
Phase I studies are usually the bridge from experiments in animal models to human subjects. For drugs, these studies are usually conducted with healthy volunteers, although they may be conducted with patients to whom the drug may be of benefit or for whom conventional therapies have failed. Phase I studies are usually not randomized or controlled (no comparison group), are small in scale, and are aimed at determining short-term safety and tolerability, dosing (including the maximally tolerated dose and toxicity) and administration through pharmacodynamic and pharmacokinetic testing, and preliminary results regarding effectiveness.
Phase II studies are based on the results from phase I studies, and are aimed at determining whether or not the intervention will have an effect on the outcomes of interest in the population to whom the intervention will be applied in clinical practice. Usually these are smaller randomized and controlled clinical trials, and the primary outcomes may be more mechanistic in nature. Phase II studies also may lead to refinements in dosing and administration, and determine short-term adverse effects and risks beyond those noted in the Phase I preliminary studies. Phase II studies will help to determine feasibility and to provide important information about assumptions around the hypothesis that will be tested in Phase III studies.
Phase III studies are aimed at providing robust evidence for clinical efficacy or effectiveness, while further specifying safety. These studies are meant to satisfy regulatory requirements for industry prior to marketing a drug or device, and are used to inform clinical recommendations and evidence-based practice. Phase III studies are rigorous randomized controlled clinical trials with large numbers of subjects, and tend to have a shorter intervention or follow-up period than would be used in clinical practice. These trials are powered to detect a clinically meaningful impact on an outcome that is immediately relevant to the patient, such as mortality, symptomatic morbidity, and functional health or quality of life. These outcomes are much harder to study in children and congenital heart patients, where the outcomes occurring are less frequent, occur over a longer period of time, or are less well-conceptualized.
Phase IV studies are long-term studies or surveillances conducted after the drug or device has received regulatory approval and is being marketed. They are aimed at monitoring the incidence of adverse events, particularly those that are rare, and to determine long-term effectiveness and safety. Phase IV studies are usually registries, although they may include clinical trials, particularly if they are aimed at determining efficacy, effectiveness, and safety for more specific indications or in more specialized populations (such as children or congenital heart patients) than in the Phase III trials.
Questions, Hypotheses, and Aims
The conceptualization of all clinical trials begins with a question. The specification of that question is founded on a sound knowledge base and preliminary evidence that leads one to a specific and focused area of controversy or uncertainty. A focused question will define the study population, the intervention and comparison, and the primary outcome for the clinical trial. The process of specification of the question forms the rationale for the clinical trial that is proposed.
The rationale informs the background/preliminary studies section of study protocols and the introduction section of manuscripts. The rationale is not merely an exhaustive literature review, but should represent a synthesis, and like any good story, should have a climax and a point. A well-conceived background to a proposal also represents the starting point for the discussion section of a manuscript once the study results are determined. In outlining these sections, one usually begins by defining the broad topic area, and then hones down to the specific area of controversy or uncertainty. The rationale is provided from both the published literature and preliminary work by the investigators. The rationale is based on the identification of deficiencies in previous studies, conflicting or paradoxical results, gaps in knowledge, or gaps in evidence. The rationale should lead directly to a statement of the research question (stated as a question), which leads to a statement of the primary aim of the trial (the question rephrased as an action item).
The relevance and significance of the question being pursued is another important aspect of the rationale. It defines the importance of the question, which is dependent on the importance of a truthful answer. Put simply, the potential answer must pass the “So what?” test. There are many dimensions to relevance and significance, which can be qualitatively or quantitatively supported. The answer may provide evidence leading to further lines of inquiry, may impact the care of patients, leading to reductions in disease burden or risk, or improvements in patient level outcomes, or may inform clinical recommendations, guidelines, or health policy. The answer should be both interesting and novel.
A hypothesis is the best guess as to what the answer to the study question might be. It should ideally be based on knowledge of the published literature and preliminary work and, hence, be an informed guess. The hypothesis should be specified at the same time that one poses the study question, which should be before any data are collected. It should also be detailed and specific to the study subjects, the intervention, and the direction and magnitude of effect on the primary outcome. Hypotheses should similarly be prespecified for each secondary research question. For the purposes of sample size estimation and inferential statistical testing, the hypotheses must be restated in a different manner, to be discussed in a later section of this chapter.
Study aims represent the main and secondary study questions, but are rephrased as an action item. As such, they are like mission statements, which define the direction or design of the clinical trial. The primary study aim should include the defining characteristic of the study population, the intervention and comparison being studied, and the primary outcome (the main or most important outcome). The design of the trial should be specific to achieve this primary aim. Sample size calculations to estimate the number of subjects required are based on the primary aim and an associated hypothesis, together with statistical considerations specifying the risk tolerance for random errors. Secondary aims are usually structured around subgroup comparisons, other types of outcomes, and mechanistic explorations. Secondary aims and hypotheses should be relevant to the primary aim, but provide a greater degree of breadth and depth to the study, which can increase the relevance and significance of a trial. One does not usually hinge sample size calculations on these secondary aims and hypotheses, but one can calculate the degree of power that may be available to test the hypotheses reliably.
Primary and secondary outcomes should be clearly specified and justified and relate directly to the study aims and underlying research questions. In the design phase of clinical trial development, much consideration is usually given to choosing the primary outcome. Preference is usually given to outcomes that are of direct relevance to the patient, with mortality being the outcome of utmost importance. Other relevant outcomes might be morbidities that impact on patient well-being, risk of intervention or further reintervention, and, more recently, assessments of functional health status or health-related quality of life. A hypothesis regarding the differential impact of the study interventions on the primary outcome informs the sample size calculations specifying the number of subjects required. Secondary outcomes are outcomes of interest that may be related to the primary outcome, either by association or informing a mechanistic relationship. The most common secondary outcomes are measures of adverse effects or events, pathophysiologic parameters, or healthcare system factors, such as utilization or costs.
Interventions and Clinical Trial Designs
The choice of intervention(s) and comparison should be evident from specification of the research question and primary aim, as well as the phase of investigations as described previously. All aspects of the intervention and its application (what, how, when, where, who) should be considered and exactly specified in advance. In pediatric trials, particular attention should be given to dosing and formulation,
and to monitoring for adverse effects. The strategy for ensuring and monitoring of compliance should be detailed. The specification of the interventions should be such that they can be replicated and applied in further studies, adapted to different populations and scenarios, or implemented in clinical practice. These details should be provided in the study protocol, the manual of operations, and in any subsequent publication of the methods and/or the results of the trial.
and to monitoring for adverse effects. The strategy for ensuring and monitoring of compliance should be detailed. The specification of the interventions should be such that they can be replicated and applied in further studies, adapted to different populations and scenarios, or implemented in clinical practice. These details should be provided in the study protocol, the manual of operations, and in any subsequent publication of the methods and/or the results of the trial.
Choice of the comparison group can be a key factor in the design of clinical trials. If aspects of the application of the intervention are likely to be influencers of outcomes or to unblind the allocation assignment, then precautions should be taken to have as many aspects as possible replicated identically for the comparison. This may include identical formulation and application of study drug and placebo, or the use of sham procedures. Often, particularly for pediatric patients, the undue burden of taking placebo or undergoing sham procedures cannot be supported due to ethical concerns. Additionally, there may be ethical concerns if there is a reasonable standard of care or alternative that may be withheld. These considerations become key drivers in the design of clinical trials in pediatrics.
The approach or perspective of the clinical trial is linked to the primary aims. Some clinical trials aim to determine if one intervention is better or safer than either usual or standard care, placebo or nonintervention, or an alternative study intervention (superiority trial). While the majority of clinical trials are undertaken with the hypothesis that the intervention of interest will be shown to be better (a one-tailed approach), statistical testing of that hypothesis is conventionally performed with a supposition that the intervention of interest may also be shown to be worse (a two-tailed approach). The two-tailed approach underlies inferential statistical testing, but it also has merit in that many interventions supported by observational studies and preliminary data are subsequently shown to be either equivalent or inferior to their comparison in a large-scale phase III trial. Alternatively and less frequently, some clinical trials aim to determine whether one intervention is no better or worse than either usual care or an alternative study intervention (noninferiority or equivalence trial). These trials are relevant when a new intervention has advantages over an established or alternative intervention (reduced costs, greater availability, improved feasibility, better tolerance, and acceptability) but there is a need to establish that outcomes will be at least equivalent and noninferior. The threshold for defining noninferior depends on the advantages of the new intervention, and there may be a trade-off for accepting some decrease in efficacy.
Simple randomized clinical trial, also called parallel group design, is the most common clinical trial design. It is characterized by subjects being recruited and randomized concurrently to only one of either an intervention or comparison, and followed concurrently to a specified single study end point. This design is the easiest to implement and to critically appraise, and is also somewhat flexible. It is limited in that it only allows one to study the relative effect of one intervention at a time.
Crossover clinical trials are a variation of the parallel group design whereby subjects are first randomized to either the intervention or comparison for a specified period of time, and then reassigned to the opposite of the initial allocation for a second specified period of time. It allows each subject to receive both intervention and comparison in randomized order. It is often used when the number of study subjects is limited and when the outcomes occur over a relatively short period of time. It allows both within-group and between-group comparisons, with each subject acting as their own control or comparison, which also helps to minimize bias and allows paired analyses. Besides increased logistical issues, one of the major limitations of this design is the problem of dropouts, since the subjects are followed longer, have increased exposure to adverse effects, and may not wish to continue with a second period of possible placebo or a less-desirable alternative. Another limitation is the potential for carryover effects, whereby the effects of the initial intervention continue to operate over the second period to influence outcomes. One method to help prevent carryover effects is to have an appropriate washout period between study periods, although this may be ineffective and further lengthen the study and contribute to dropout. Both of these limitations complicate the analysis and interpretation of the trial results.
A factorial design allows the study of more than one intervention together (interaction) as well as separately (independent). The least complex factorial design randomizes subjects to one of four groups—neither intervention (comparison), both interventions, or each intervention separately—and is called a 2 × 2 factorial design. This design can be efficient in determining both the individual and combined benefits of interventions, including synergy, but also carries the risk of unexpected adverse effects from the combination. Factorial design trials require larger numbers of subjects and have greater logistical issues, and the analysis and interpretation can be complex.
Study Subjects
The degree to which the study subjects that enroll and complete the trial are representative of all applicable patients, meaning similar in characteristics and response, depends on how the subjects were chosen, recruited and maintained as participants in the trial. These aspects can have important impacts on the validity of the results and their generalizability, and are outlined in Table 81.7.
TABLE 81.7 Defining and Selecting the Study Subjects | |
|---|---|
|
Definition of the study population begins with examination of the aims and hypotheses of the clinical trial. One wishes to infer that the results of the trial for those subjects studied will be representative of the truth if the trial had been performed in the universe of potential subjects, or the target population. It is usually given that one cannot study absolutely everyone, but one can study those potential subjects who can be identified and approached by the investigators, or the accessible population. Of the accessible population, some subjects cannot be contacted, will prove not appropriate for study, may decline to participate or may start but not complete their participation, and represent the actual study subjects. The degree to which, at each stage, the subjects are similar to the target population will inform the validity of inferences based on the results from the actual study subjects.
Inclusion and exclusion criteria are defined to refine specification of the accessible population that may be recruited for study participation. Inclusion criteria are the defining characteristics specific to the target population applicable to the study aims. They usually include demographic characteristics (age, sex), clinical characteristics (anatomy, diagnoses, morbidities, procedures), accessibility characteristics (geography, settings), and the time period. Exclusion criteria are characteristics that may influence participation (language barriers,
anticipated poor compliance or follow-up, cognitive or physical limitations), applicability or safety of the study interventions (contraindications), or unduly influence outcomes (associated conditions or treatments). Each criterion should be exactly and objectively defined as far as possible. There must be an appropriate balance between being too general and too overly restrictive, balancing generalizability with feasibility.
anticipated poor compliance or follow-up, cognitive or physical limitations), applicability or safety of the study interventions (contraindications), or unduly influence outcomes (associated conditions or treatments). Each criterion should be exactly and objectively defined as far as possible. There must be an appropriate balance between being too general and too overly restrictive, balancing generalizability with feasibility.
Sampling strategies may sometimes be necessary when the number of accessible and potentially eligible subjects exceeds the number of subjects required, but one wishes to minimize selection bias. Convenience sampling is most liable to potential bias, and involves selecting subjects who are the most accessible, such as enrolling subjects as they present from routinely scheduled clinic visits. Systematic sampling involves having a system to select subjects, such as enrolling no more than four subjects per week from the surgical list. Simple random sampling involves creating a list of all accessible subjects and then selecting subjects at random until the desired number is achieved, and is the method most likely to minimize selection bias. Random sampling can be stratified by dividing the accessible subjects into defined subgroups, and then sampling within those subgroups, and may be used when the investigators wish to examine the differential effects of the study interventions. If used, any sampling strategy must be exactly specified and tamperproof.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


