Chapter 44 Cystic Fibrosis
Genetics
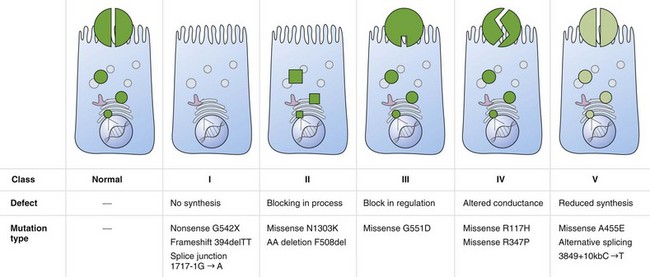
Cystic fibrosis is caused by mutations in a gene on chromosome 7 encoding the protein subsequently termed the CFTR gene. More than 1800 mutations have been reported to the Cystic Fibrosis Genetic Analysis Consortium. Most of these mutations are rare, and only four mutations occur in a frequency of more than 1%. CFTR mutations can be grouped into five classes: CFTR is not synthesized (I), is inadequately processed (II), is not regulated (III), shows abnormal conductance (IV), or has partially defective production or processing (V). Class I, II, and III mutations are more common and associated with pancreatic insufficiency, whereas patients with the less common class IV and V mutations often are “pancreatic sufficient” (Figure 44-1).
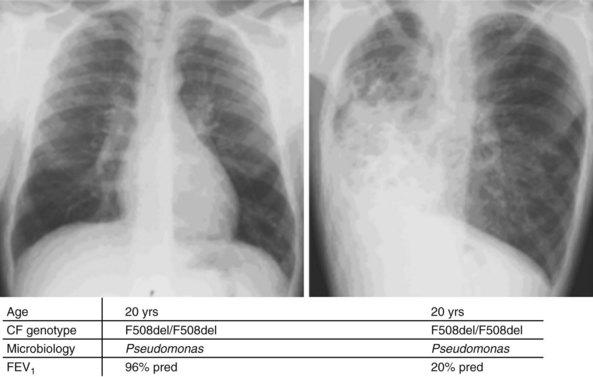
Whereas classes IV and V CFTR mutations are linked with pancreatic sufficiency, attempts to link specific mutations to the severity of lung disease have shown large phenotypic variability. This is best documented for patients homozygous for the F508del mutation who exhibit a wide spectrum in lung disease severity. This wide phenotypic variation suggests that environmental factors and genes other than CFTR influence the development, progression, and disease severity of CF (Figure 44-2).
Pathophysiology
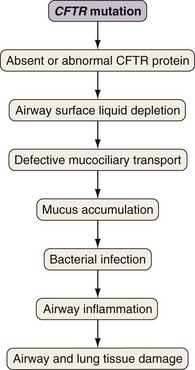
Airway epithelial cells secrete chloride and absorb sodium chloride (NaCl), the balance of which is regulated through apical channels, including CFTR (Figure 44-3). Ion secretion and absorption affect water transport, and a balance between secretion and absorption is thought to be important to maintain an adequate layer of airway surface liquid (ASL). The ASL supports the thin mucous layer on top of epithelial cells, which is constantly transported out of the lungs through ciliary movement. Lack or dysfunction of CFTR leads to reduced chloride secretion and NaCl hyperabsorption with depletion of ASL. In the absence of adequate ASL, respiratory cilia collapse, leading to breakdown of mucociliary transport. Mucus accumulates in the lower airways, and inhaled bacteria are trapped in this viscous mucous layer on top of respiratory epithelial cells.
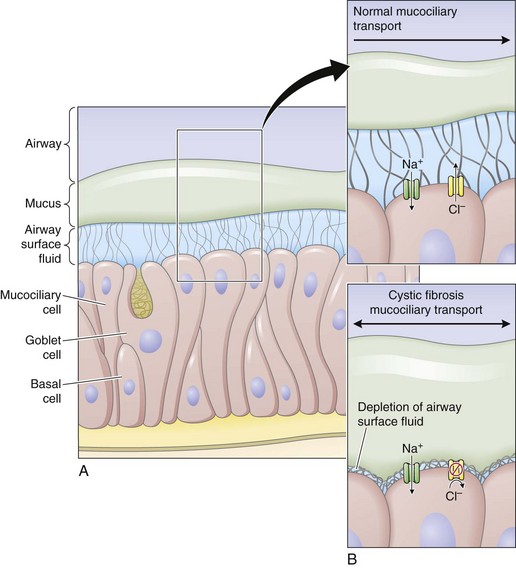
Figure 44-3 Restoring airway surface liquid in cystic fibrosis.
(From Ratjen F: Restoring airway surface liquid in cystic fibrosis, N Engl J Med 354:291–293, 2006.)
The spectrum of bacteria that are relevant for CF lung disease is relatively limited. Overall, Pseudomonas aeruginosa is the most common isolate, followed by Staphylococcus aureus and Haemophilus influenzae. Later in the course of disease, multiresistant organisms such as Stenotrophomonas maltophilia, Achromobacter (Alcaligenes) xylosoxidans, and Burkholderia cepacia complex may be isolated. As in other chronic pulmonary diseases, nontuberculous mycobacteria (usually Mycobacterium avium-intracellulare or M. abscessus) may be isolated. It is challenging to prove whether these organisms are causing ongoing disease requiring treatment, or if they are colonizing only the damaged lung. For a more detailed discussion of nontuberculous mycobacteria (NTM) infections, see Chapter 31.
Evidence indicates that inflammation is dysregulated in CF airways. Neutrophilic airway inflammation has been detected in infants with CF in the first months of life, as well as in CF fetal lung tissue. Whether or not inflammation is directly related to the CFTR defect is still disputed. However, an exaggerated, sustained, and prolonged inflammatory response to bacterial and viral pathogens is an accepted feature of CF lung disease. The persistent endobronchial inflammation is deleterious for the course of lung disease (Figure 44-4).
Clinical Features
Typical signs and symptoms for CF are listed in Box 44-1. Symptoms of CF may vary, with monosymptomatic cases often diagnosed late. It is therefore important to be aware of the spectrum of symptoms that may arise and to initiate adequate diagnostic steps.
Diagnostic Approach
The diagnosis of cystic fibrosis is established by clinical manifestations (see Box 44-1), a history of CF in a sibling, or a positive newborn screening result, in conjunction with laboratory evidence of CFTR dysfunction. CFTR dysfunction is documented by elevated sweat chloride or characteristic abnormalities in nasal potential difference or by CF-causing mutations in the CFTR gene.
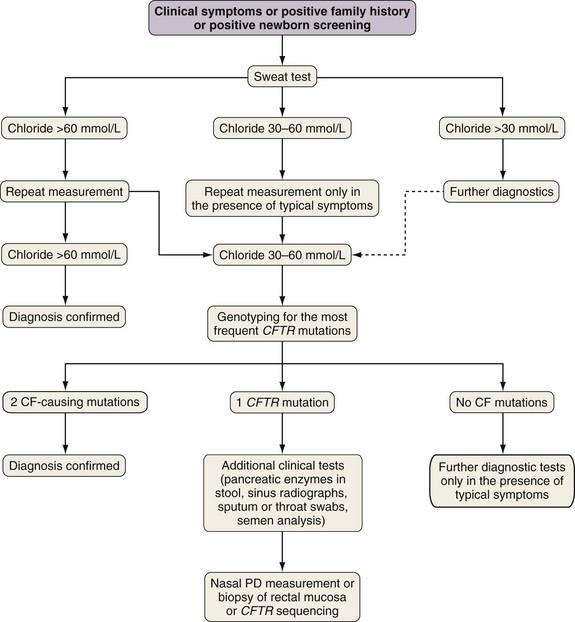
A diagnostic algorithm is presented in Figure 44-5. Abnormal ion transport is reflected in high sweat NaCl levels, and measurement of chloride concentration in sweat after iontophoresis of pilocarpine is used for diagnosis. Sweat testing must be done using standardized methods, by qualified staff in an experienced laboratory. A sweat chloride concentration greater than 60 mmol/L on repeated analysis is diagnostic for CF; 30 to 60 mmol/L is considered a “borderline” result but may be seen in patients with CF.