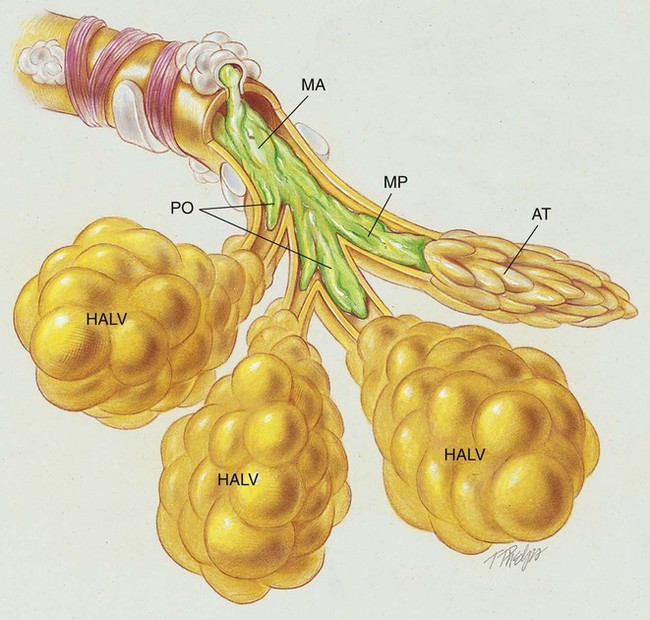
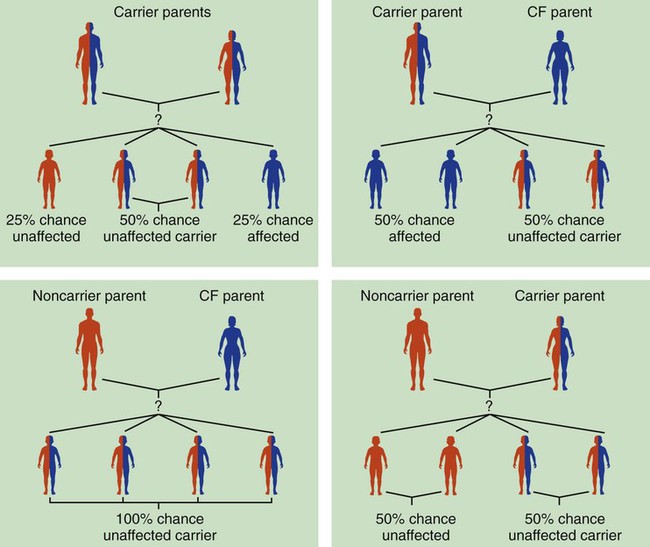
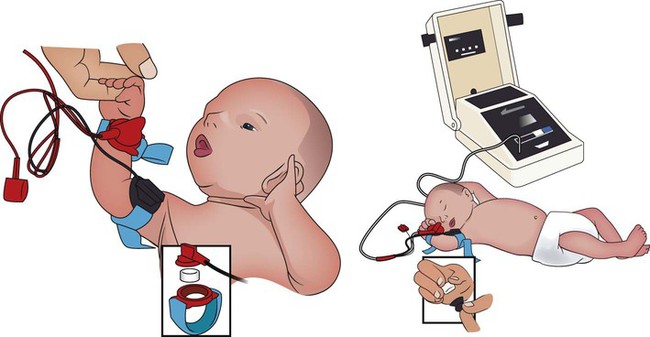
After reading this chapter, you will be able to: • List the anatomic alterations of the lungs associated with cystic fibrosis. • Describe the causes and classifications of cystic fibrosis. • List the cardiopulmonary clinical manifestations associated with cystic fibrosis. • Describe the general management of cystic fibrosis. • Describe the clinical strategies and rationales of the SOAPs presented in the case study. • Define key terms and complete self-assessment questions at the end of the chapter and on Evolve. The abundance of stagnant mucus in the tracheobronchial tree also serves as an excellent culture medium for bacteria, particularly Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa. Some gram-negative bacteria are also commonly associated with cystic fibrosis, such as Stenotrophomonas maltophilia, Burkholderia cepacia, Burkholderia pickettii, and Burkholderia gladioli. The infection stimulates additional mucous production and further compromises the mucociliary transport system. This condition may lead to secondary bronchial smooth muscle constriction. Finally, as the disease progresses, the patient may develop signs and symptoms of recurrent pneumonia, chronic bronchitis, bronchiectasis, and lung abscesses (Figure 14-1). The major pathologic or structural changes associated with cystic fibrosis are as follows: Because cystic fibrosis is a recessive gene disorder, the child must inherit two copies of the defective cystic fibrosis gene—one from each parent (cystic fibrosis carriers)—to have the disease. Even though the carrier of the cystic fibrosis gene may be identified through genetic testing, the carrier (heterozygote) does not demonstrate evidence of the disease. However, if both parents carry the cystic fibrosis gene, the possibility of their children having cystic fibrosis (regardless of gender) follows the standard Mendelian pattern: there is a 25% chance that each child will have cystic fibrosis, a 25% chance that each child will be completely normal (and not carry the gene), and a 50% chance that each child will be a carrier. Thus, when both patients have the cystic fibrosis gene, there is a one in four chance that the child will have cystic fibrosis (Figure 14-2). It is estimated that more than 10 million Americans are unknowing, symptomless carriers of the mutant cystic fibrosis gene. According to the Cystic Fibrosis Foundation, cystic fibrosis affects about 30,000 children and adults in the United States. About 1000 new cases of cystic fibrosis are diagnosed each year in the United States (70,000 worldwide). More than 70% of the patients are diagnosed by age 2. More than 40% of the cystic fibrosis patient population is age 18 or older.* Other researchers state that whites are most often affected (1 in 2500 to 3500). Cystic fibrosis is less common in Hispanics (1 in 9500) and African-Americans (1 in 17,000). Cystic fibrosis is rarely seen in Asians (1 in 31,000). The predicted median life expectancy is 37 years. Death usually is caused by pulmonary complications. The diagnosis of cystic fibrosis is based on clinical manifestations associated with cystic fibrosis, family history of cystic fibrosis, and laboratory findings. Box 14-1 provides common clinical indicators that justify evaluation for cystic fibrosis. The sweat test (sometimes called the sweat chlorine test) is the gold standard diagnostic test for cystic fibrosis. The sweat test is a reliable test for the identification of approximately 98% of patients with cystic fibrosis. This test measures the amount of sodium and chloride in the patient’s sweat. During the procedure a small amount of a colorless, odorless sweat-producing chemical called pilocarpine is applied to the patient’s arm or leg—usually the forearm. An electrode is attached to the chemically prepared area, and a mild electric current is applied to stimulate sweat production (Figure 14-3). To collect the sweat, the area is covered with a gauze pad or filter paper and wrapped in plastic. After about 30 minutes the plastic is removed, and the sweat collected in the pad or paper is sent to the laboratory for analysis. The test is usually done twice. • Amniocentesis—the amniotic fluid is obtained and tested to determine if both of the CFTR genes from the fetus are normal. Amniocentesis can be used to diagnose a large number of genetic and chromosomal abnormalities, including cystic fibrosis. • Chorionic villus biopsy—with the aid of an ultrasound examination, a thin tube is inserted into the uterus to obtain a piece of the placenta to biopsy. The cells of the placenta are then tested for a variety of genetic defects, including cystic fibrosis. The following clinical manifestations result from the pathophysiologic mechanisms caused (or activated) by Atelectasis (see Figure 9-8), Bronchospasm (see Figure 9-11), and Excessive Bronchial Secretions (see Figure 9-12)—the major anatomic alterations of the lungs associated with cystic fibrosis (CF) (see Figure 14-1).
Cystic Fibrosis
Anatomic Alterations of the Lungs*
Etiology and Epidemiology
How the Cystic Fibrosis Gene Is Inherited
Screening and Diagnosis
Sweat Test
Prenatal Testing
 OVERVIEW of the Cardiopulmonary Clinical Manifestations Associated with Cystic Fibrosis
OVERVIEW of the Cardiopulmonary Clinical Manifestations Associated with Cystic Fibrosis