Elliott M. Antman
Critical Evaluation of Clinical Trials
Despite many decades of advances in diagnosis and management, cardiovascular disease (CVD) remains the leading cause of death in the United States and other high-income countries, as well as many developing countries.1 Managing the burden of CVD consumes 16% of overall national health care expenditures in the United States; interventions to treat CVD are therefore a major focus of contemporary clinical research. Therapeutic recommendations are no longer based on nonquantitative pathophysiologic reasoning but instead are evidence-based. Rigorously performed trials are required before regulatory approval and clinical acceptance of new treatments (drugs, devices, and biologics) and biomarkers.2 Thus the design, conduct, analysis, interpretation, and presentation of clinical trials constitute a central feature of the professional life of the contemporary cardiovascular specialist.3,4 Case-control studies and analyses from registries are integral to epidemiologic and outcomes research but are not strictly clinical trials and are not discussed in this chapter.5,6
Constructing the Research Question
Before embarking on a clinical trial, investigators should review the FINER criteria for a good research question (Table 6-1) and the phases of evaluation of new therapies (Table 6-2) and should familiarize themselves with the processes of designing and implementing a research project, good clinical practice, and drawing conclusions from the findings ( Fig. e6-1).3,4,6–10 A clinical trial may be designed to test for superiority of the investigational treatment over the control therapy but also may be designed to show therapeutic similarity between the investigational and the control treatments (noninferiority design) (Fig. 6-1; Table 6-3).
Fig. e6-1).3,4,6–10 A clinical trial may be designed to test for superiority of the investigational treatment over the control therapy but also may be designed to show therapeutic similarity between the investigational and the control treatments (noninferiority design) (Fig. 6-1; Table 6-3).
TABLE 6-1
FINER Criteria for a Good Research Question
| F | Feasible |
| I | Interesting |
| N | Novel |
| E | Ethical |
| R | Relevant |
From Hulley SB, Cummings SF, Browner WS, et al: Designing Clinical Research. 3rd ed. Philadelphia, Lippincott Williams & Wilkins, 2007.
TABLE 6-2
Phases of Evaluations of New Therapies
| PHASE | FEATURES | PURPOSE |
| I | First administration of new treatment | Safety—is further investigation warranted? |
| II | Early trial in patients | Efficacy—dose ranging, adverse events, pathophysiologic insights |
| III | Large scale comparison versus standard treatment | Registration pathway—definitive evaluation |
| IV | Monitoring in clinical practice | Postmarketing surveillance |
Modified from Meinert C: Clinical trials. Design, conduct, and analysis. New York, Oxford University Press, 1986; and Stanley K: Design of randomized controlled trials. Circulation 115:1164, 2007.
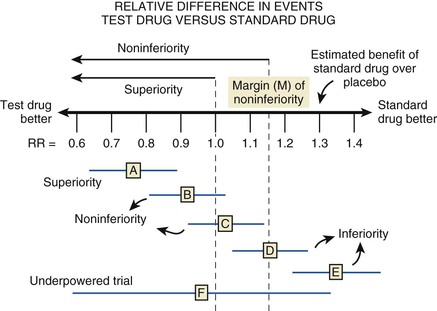
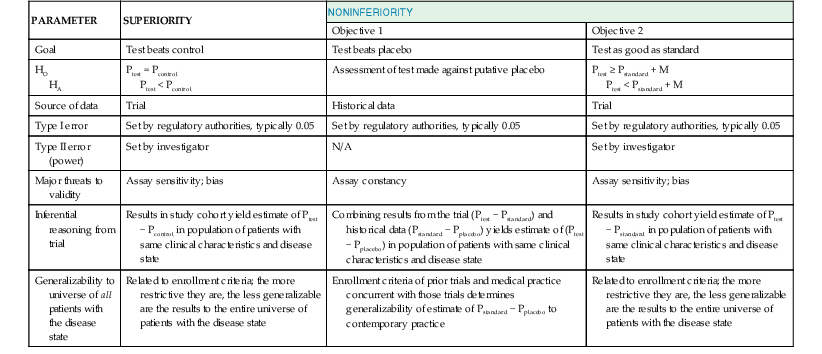
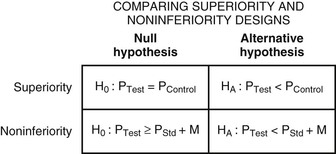
Regardless of the design of the trial, it is essential that investigators provide a statement of the hypothesis being examined, using a format that permits biostatistical assessment of the results (see Fig. e6-1). Typically, a null hypothesis (H0) is specified (e.g., no difference exists between the treatments being studied) and the trial is designed to provide evidence leading to rejection of H0 in favor of an alternative hypothesis (HA) (a difference exists between treatments). To determine whether H0 may be rejected, investigators specify type I (α) and type II (β) errors, referred to as the false-positive and false-negative rates, respectively. By convention, α is set at 5%, indicating a willingness to accept a 5% probability that a significant difference will occur by chance when there is no true difference in efficacy. Regulatory authorities may on occasion demand a more stringent level of α—for example, when a single large trial is being proposed rather than two smaller trials—to gain approval of a new treatment. The value of β represents the probability that a specific difference in treatment efficacy might be missed, so that the investigators incorrectly fail to reject H0 when there is a true difference in efficacy. The power of the trial is given by the quantity (1 − β) and is selected by the investigators—typically, between 80% and 90%.7 Using the quantities α, β, and the estimated event rates in the control group, the sample size of the trial can be calculated with formulas for comparison of dichotomous outcomes or for a comparison of the rate of development of events over a follow-up period (time to failure). Table 6-3 summarizes the major features and concepts for superiority and noninferiority trials designed to change the standard of care for patients with a cardiovascular condition.
Clinical Trial Design
Controlled Trials
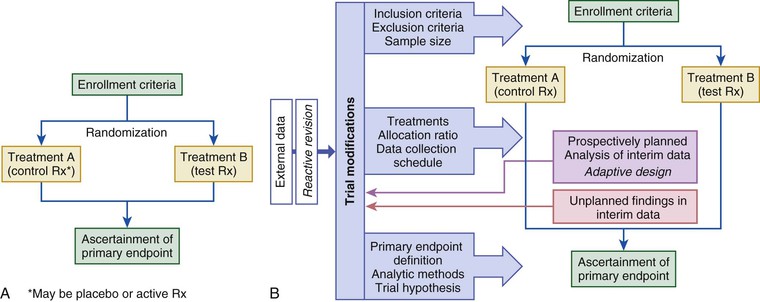
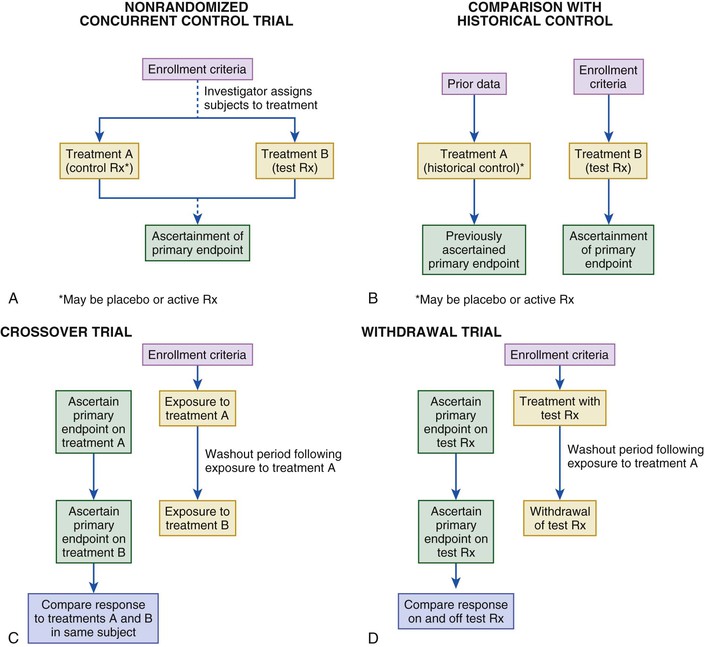
The randomized controlled trial (RCT) is considered the gold standard for the evaluation of new treatments (Fig. 6-2). The allocation of subjects to control and test treatments is not determined but is based on an impartial scheme (usually a computer algorithm). Randomization reduces the likelihood of patient selection bias in allocation of treatment, enhances the likelihood that any baseline differences between groups are random so that comparable groups of subjects can be compared, and validates the use of common statistical tests. Randomization may be fixed over the course of the trial or may be adaptive, based on the distribution of treatment assignments in the trial to a given point, baseline characteristics, or observed outcomes (see Fig. 6-2A).15 Fixed randomization schemes are more common and are specified further according to the allocation ratio (equal or unequal assignment to study groups), stratification levels, and block size (i.e., constraining the randomization of patients to ensure a balanced number of assignments to the study groups, especially if stratification [e.g., based on enrollment characteristics] is used in the trial). During the course of a trial, investigators may find it necessary to modify one or more treatments in response to evolving data (internal or external to the trial) or a recommendation from the trial’s data safety monitoring board (DSMB)—that is, to implement an adaptive design (see Fig. 6-2B).15 Adaptive designs are most readily implemented during phase II of therapeutic development. Regulatory authorities are concerned about protection of the trial integrity and the studywise alpha level when adaptive designs are used in registration pathway trials.15 The most desirable situation is for the control group to be studied concurrently and to comprise subjects distinct from those of the treatment group. Other trial formats that have been used in cardiovascular investigations include nonrandomized concurrent and historical controls (Fig. 6-3A, B), crossover designs (see Fig. 6-3C), withdrawal trials (see Fig. 6-3D), and group or cluster allocations (groups of subjects or investigative sites are assigned as a block to test or control). Depending on the clinical circumstances, the control agent may be a placebo or a drug or other intervention used in active treatment (standard of care).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


