John M. Canty Jr.,, Dirk J. Duncker
Coronary Blood Flow and Myocardial Ischemia
The coronary circulation is unique in that the heart is responsible for generating the arterial pressure that is required to perfuse the systemic circulation and yet, at the same time, has its own perfusion impeded during the systolic portion of the cardiac cycle. Because myocardial contraction is closely connected to coronary flow and oxygen delivery, the balance between oxygen supply and demand is a critical determinant of the normal beat-to-beat function of the heart (Feigl, 1983 [classic reading]). When this relation is acutely disrupted by diseases affecting coronary blood flow the resulting imbalance can immediately precipitate a vicious cycle, whereby ischemia-induced contractile dysfunction precipitates hypotension and further myocardial ischemia. Thus knowledge of the regulation of coronary blood flow, determinants of myocardial oxygen consumption, and the relation between ischemia and contraction is essential for understanding the pathophysiologic basis and management of many cardiovascular disorders (Hoffman and Spaan, 1990 [classic reading]).
Control of Coronary Blood Flow
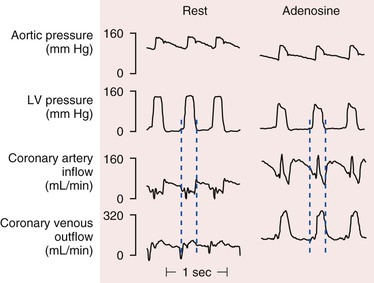
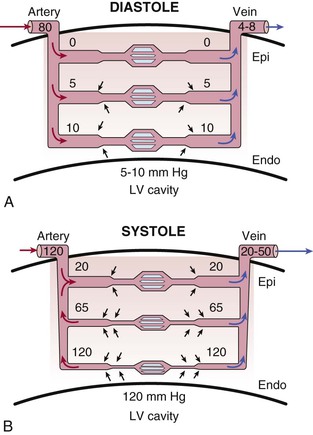
There are pronounced systolic and diastolic coronary flow variations throughout the cardiac cycle with coronary arterial inflow out of phase with venous outflow (Fig. 49-1) Systolic contraction increases tissue pressure, redistributes perfusion from the subendocardial to the subepicardial layers of the heart, and impedes coronary arterial inflow, which reaches a nadir. At the same time, systolic compression reduces the diameter of intramyocardial microcirculatory vessels (arterioles, capillaries, and venules) and increases coronary venous outflow, which peaks during systole. During diastole, coronary arterial inflow increases with a transmural gradient that favors perfusion to the subendocardial vessels. At this time, coronary venous outflow falls.
Determinants of Myocardial Oxygen Consumption
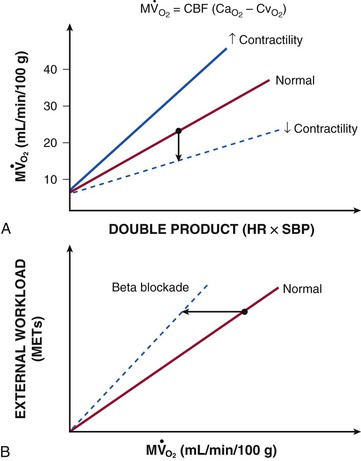
In contrast to most other vascular beds, myocardial oxygen extraction is near-maximal at rest, averaging 60% to 80% of arterial oxygen content.1 The ability to increase oxygen extraction as a means to increase oxygen delivery is limited to circumstances associated with sympathetic activation and acute subendocardial ischemia. Nevertheless, coronary venous oxygen tension (PVo2) can only decrease from 25 mm Hg to approximately 15 mm Hg. Because of the high resting oxygen extraction, increases in myocardial oxygen consumption are primarily met by proportional increases in coronary flow and oxygen delivery (Fig. 49-2). In addition to coronary flow, oxygen delivery is directly determined by arterial oxygen content (Cao2). This is equal to the product of hemoglobin concentration and arterial oxygen saturation plus a small amount of oxygen dissolved in plasma that is directly related to arterial oxygen tension (Pao2). Thus, for any given flow level, anemia results in proportional reductions in oxygen delivery whereas hypoxia, due to the nonlinear oxygen dissociation curve, results in relatively small reductions in oxygen content until Pao2 falls to the steep portion of the oxygen dissociation curve (below 50 mm Hg).
 ).
).  are primarily met by increases in coronary flow and linearly related to the double product. A doubling of HR, SBP, or contractility each results in approximately 50% increases in myocardial oxygen consumption.
are primarily met by increases in coronary flow and linearly related to the double product. A doubling of HR, SBP, or contractility each results in approximately 50% increases in myocardial oxygen consumption. 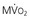 ) by reducing the double product and myocardial contractility. Ca
) by reducing the double product and myocardial contractility. CaThe major determinants of myocardial oxygen consumption are heart rate, systolic pressure (or myocardial wall stress), and left ventricular (LV) contractility (see Figs. 21-21 and 54-2). A twofold increase in any of these individual determinants of oxygen consumption requires an approximately 50% increase in coronary flow. Experimentally, the systolic pressure volume area is proportional to myocardial work and linearly related to myocardial oxygen consumption. The basal myocardial oxygen requirements needed to maintain critical membrane function are low (approximately 15% of resting oxygen consumption), and the cost of electrical activation is trivial when mechanical contraction ceases during diastolic arrest (as with cardioplegia) and diminishes during ischemia.
Coronary Autoregulation
Regional coronary blood flow remains constant as coronary artery pressure is reduced below aortic pressure over a wide range when the determinants of myocardial oxygen consumption are kept constant. This phenomenon is termed autoregulation (Fig. 49-3). When pressure falls to the lower limit of autoregulation, coronary resistance arteries are maximally vasodilated to intrinsic stimuli, and flow becomes pressure-dependent, resulting in the onset of subendocardial ischemia. Resting coronary blood flow under normal hemodynamic conditions averages 0.7 to 1.0 mL/min/g and can increase between four- and fivefold during vasodilation (Klocke, 1976 [classic reading]). The ability to increase flow above resting values in response to pharmacologic vasodilation is termed coronary flow reserve. Flow in the maximally vasodilated heart is dependent on coronary arterial pressure. Maximum perfusion and coronary flow reserve are reduced when the diastolic time available for subendocardial perfusion is decreased (tachycardia) or the compressive determinants of diastolic perfusion (preload) are increased. Coronary reserve also is diminished by anything that increases resting flow, including increases in the hemodynamic determinants of oxygen consumption (systolic pressure, heart rate, and contractility) and reductions in arterial oxygen supply (anemia and hypoxia). Thus circumstances can develop that precipitate subendocardial ischemia in the presence of normal coronary arteries (Hoffman and Spaan, 1990 [classic reading]). Although initial studies suggested that the lower pressure limit of autoregulation is 70 mm Hg, Canty and associates (Fig. 49-4) have shown that coronary flow can be autoregulated to mean coronary pressures as low as 40 mm Hg (diastolic pressures of 30 mm Hg) in conscious dogs in the basal state. These coronary pressure levels are similar to those recorded in humans without symptoms of ischemia, distal to chronic coronary occlusions, using pressure wire micromanometers. The lower autoregulatory pressure limit increases during tachycardia because of an increase in flow requirements, as well as a reduction in the time available for perfusion.
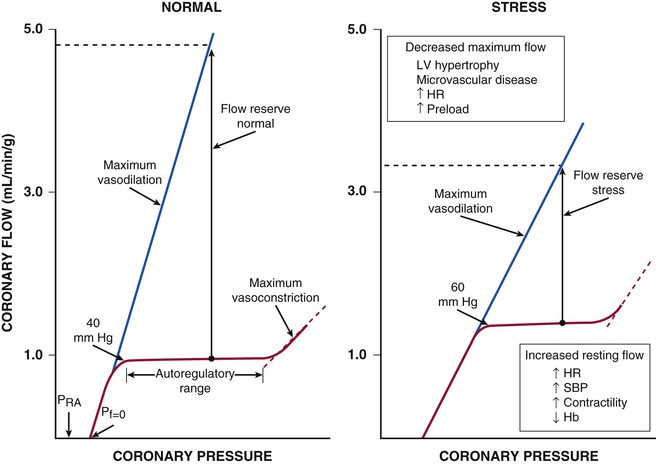
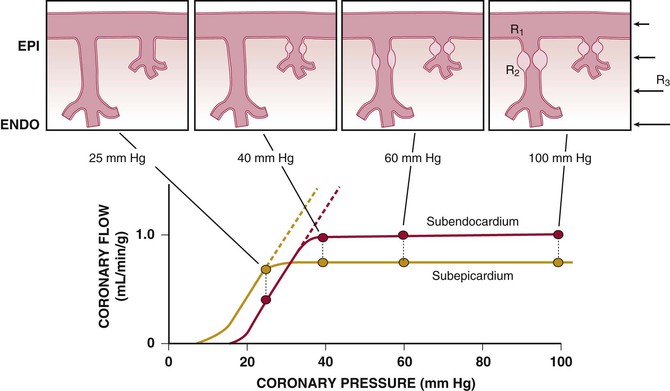
Figure 49-4 also illustrates important transmural variations in the lower autoregulatory pressure limit, which result in increased vulnerability of the subendocardium to ischemia. Subendocardial flow occurs primarily in diastole and begins to decrease below a mean coronary pressure of 40 mm Hg. By contrast, subepicardial flow occurs throughout the cardiac cycle and is maintained until coronary pressure falls below 25 mm Hg. This difference arises from increased oxygen consumption in the subendocardium, requiring a higher resting flow level, as well as the more pronounced effects of systolic contraction on subendocardial vasodilator reserve. The transmural difference in the lower autoregulatory pressure limit results in vulnerability of the subendocardium to ischemia in the presence of a coronary stenosis. Although there is no pharmacologically recruitable flow reserve during ischemia in the normal coronary circulation, reductions in coronary flow below the lower limit of autoregulation can occur in the presence of pharmacologically recruitable coronary flow reserve under certain circumstances.2
Endothelium-Dependent Modulation of Coronary Tone.
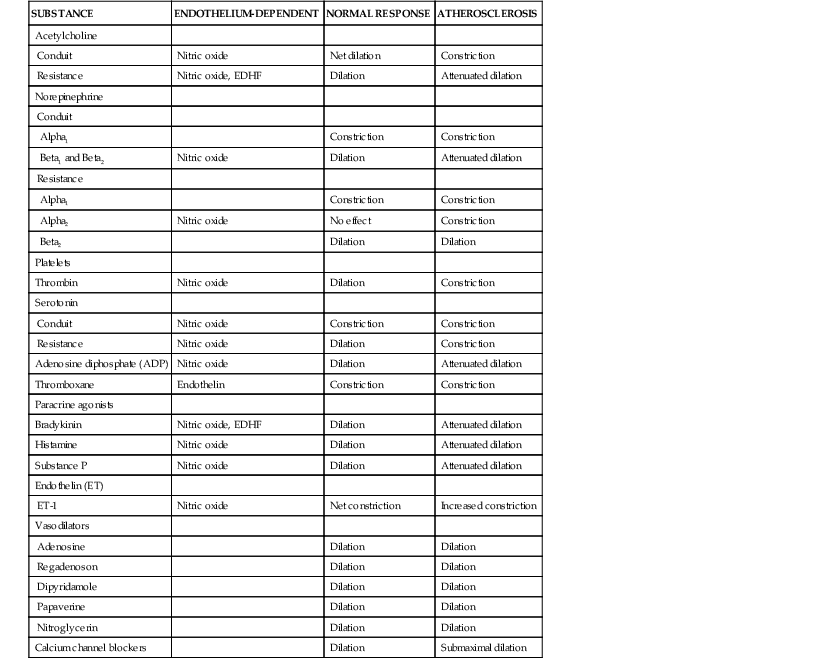
Epicardial conduit arteries do not contribute significantly to coronary vascular resistance, yet arterial diameter is modulated by a wide variety of paracrine factors that can be released from platelets, as well as circulating neurohormonal agonists, neural tone, and local control through vascular shear stress.1 The most common factors related to cardiovascular disease are summarized in Table 49-1 (see also Fig. e49-1 ). The net effect of many of these agonists is critically dependent on whether a functional endothelium is present. Furchgott and Zawadzki (1980 [classic reading]) originally demonstrated that acetylcholine normally dilates arteries via an endothelium-dependent relaxing factor that was later identified to be nitric oxide (NO). This binds to guanylyl cyclase and increases cyclic guanosine monophosphate (cGMP), resulting in vascular smooth muscle relaxation. When the endothelium is removed, the dilation to acetylcholine is converted to vasoconstriction, reflecting the effect of muscarinic vascular smooth muscle contraction. Subsequent studies have demonstrated that coronary resistance arteries also exhibit endothelial modulation of diameter and that the response to physical forces such as shear stress, as well as paracrine mediators, vary with resistance vessel size.3,4 The major endothelium-dependent biochemical pathways involved in regulating coronary epicardial and resistance artery diameter are as follows.
). The net effect of many of these agonists is critically dependent on whether a functional endothelium is present. Furchgott and Zawadzki (1980 [classic reading]) originally demonstrated that acetylcholine normally dilates arteries via an endothelium-dependent relaxing factor that was later identified to be nitric oxide (NO). This binds to guanylyl cyclase and increases cyclic guanosine monophosphate (cGMP), resulting in vascular smooth muscle relaxation. When the endothelium is removed, the dilation to acetylcholine is converted to vasoconstriction, reflecting the effect of muscarinic vascular smooth muscle contraction. Subsequent studies have demonstrated that coronary resistance arteries also exhibit endothelial modulation of diameter and that the response to physical forces such as shear stress, as well as paracrine mediators, vary with resistance vessel size.3,4 The major endothelium-dependent biochemical pathways involved in regulating coronary epicardial and resistance artery diameter are as follows.
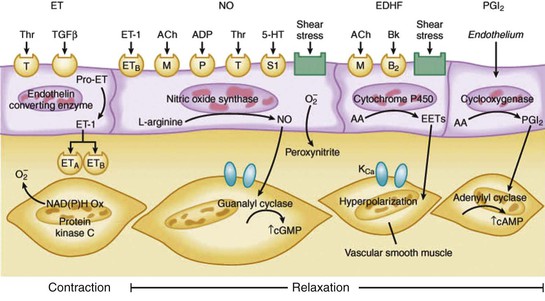
Nitric Oxide (Endothelium-Derived Relaxing Factor).
NO is produced in endothelial cells by the enzymatic conversion of l-arginine to citrulline via type III NO synthase (NOS). Endothelial NO diffuses abluminally into vascular smooth muscle, where it binds to guanylyl cyclase, increasing cGMP production and causing relaxation through a reduction in intracellular calcium. NO-mediated vasodilation is enhanced by cyclic or pulsatile changes in coronary shear stress. Chronic upregulation of NOS occurs in response to episodic increases in coronary flow, such as during exercise training, which also potentiates the relaxation to various endothelium-dependent vasodilators. NO-mediated vasodilation is impaired in many disease states and in patients with one or more risk factors for coronary artery disease (CAD). This occurs via inactivation of NO by superoxide anion generated in response to oxidative stress. Such inactivation is the hallmark of impaired NO-mediated vasodilation in atherosclerosis, hypertension, and diabetes.
Endothelium-Dependent Hyperpolarizing Factor.
Endothelium-dependent hyperpolarization is an additional endothelium-dependent mechanism for selected agonists (e.g., bradykinin), as well as shear stress–induced vasodilation, in the human coronary microcirculation. Endothelium-dependent hyperpolarizing factor (EDHF), produced by the endothelium, hyperpolarizes vascular smooth muscle and dilates arteries by opening calcium-activated potassium channels (KCa). The exact biochemical species of EDHF is still unclear, but prominent candidates are endothelium-derived hydrogen peroxide and epoxyeicosatrienoic acid, a metabolite of arachidonic acid metabolism produced by the cytochrome P-450 epoxygenase pathway.5
Prostacyclin.
Metabolism of arachidonic acid via cyclooxygenase also can produce prostacyclin, which is a coronary vasodilator when administered exogenously. Although some evidence indicates that prostacyclin contributes to tonic coronary vasodilation, inhibitors of cyclooxygenase fail to alter flow during ischemia distal to an acute stenosis or limit oxygen consumption in response to increases in metabolism. This suggests that it is overcome by other compensatory vasodilator pathways.2 In contrast with the coronary resistance vasculature, vasodilator prostaglandins are very important determinants of coronary collateral vessel resistance, and inhibiting cyclooxygenase reduces collateral perfusion in dogs.6
Endothelin.
The endothelins—ET-1, ET-2, and ET-3—are peptide endothelium-dependent constricting factors. ET-1 is a potent constrictor derived from the enzymatic cleavage of a larger precursor molecule (pre-pro–endothelin) via endothelin-converting enzyme. In contrast with the rapid vascular smooth muscle relaxation and recovery characteristic of endothelium-derived vasodilators (NO, EDHF, and prostacyclin), the constriction to endothelin is prolonged. Changes in endothelin levels are largely mediated through transcriptional control and produce longer-term changes in coronary vasomotor tone. The effects of endothelin are mediated by binding to both ETA and ETB receptors. ETA-mediated constriction is caused by the activation of protein kinase C in vascular smooth muscle. ETB-mediated constriction is less pronounced and counterbalanced by ETB-mediated endothelium-dependent NO production and vasodilation. Endothelin is only marginally involved in regulating coronary blood flow in the normal heart but can modulate vascular tone when interstitial and circulating concentrations increase in pathophysiologic states such as heart failure.
Determinants of Coronary Vascular Resistance
The resistance to coronary blood flow can be divided into three major components, as summarized in Figure 49-5 (Klocke, 1976 [classic reading]). Under normal circumstances, there is no measurable pressure drop in the epicardial arteries, indicating negligible conduit resistance (R1). With the development of hemodynamically significant epicardial artery narrowing (more than 50% diameter reduction), the fixed conduit artery resistance begins to contribute an increasing component to total coronary resistance and, when severely narrowed (more than 90%), may reduce resting flow.
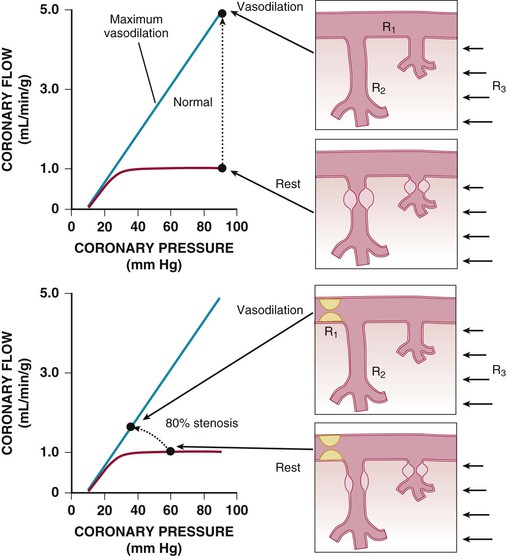
The second component of coronary resistance (R2) is dynamic and arises primarily from microcirculatory resistance arteries and arterioles. This is distributed throughout the myocardium across a broad range of microcirculatory resistance vessel sizes (20 to 400 µm in diameter) and changes in response to physical forces (intraluminal pressure and shear stress), as well as the metabolic needs of the tissue. Normally, little resistance is contributed by coronary venules and capillaries, and their resistance remains fairly constant during changes in vasomotor tone. Even in the maximally vasodilated heart, capillary resistance accounts for no more than 20% of the microvascular resistance.4 Thus a twofold increase in capillary density would increase maximal myocardial perfusion by only approximately 10%. Minimal coronary vascular resistance of the microcirculation is primarily determined by the size and density of arterial resistance vessels and results in substantial coronary flow reserve in the normal heart.
Extravascular Compressive Resistance
The third component, extravascular compressive resistance (R3), varies with time throughout the cardiac cycle and is related to cardiac contraction and systolic pressure development within the left ventricle. In heart failure, compressive effects from elevated ventricular diastolic pressure also impede perfusion by passive compression of microcirculatory vessels from elevated extravascular tissue pressure during diastole. Increases in preload effectively raise the normal back pressure to coronary flow above coronary venous pressure levels (Hoffman and Spaan, 1990 [classic reading]). Compressive effects are most prominent in the subendocardium and are discussed in greater detail further on.
During systole, cardiac contraction raises extravascular tissue pressure to values equal to LV pressure at the subendocardium. This declines to values near pleural pressure at the subepicardium. The increased effective back pressure during systole produces a time-varying reduction in the driving pressure for coronary flow that impedes perfusion to the subendocardium. Although this paradigm can explain variations in systolic coronary inflow, it is not able to account for the increase in coronary venous systolic outflow. To explain both impaired inflow and accelerated venous outflow, some investigators have proposed the concept of the intramyocardial pump (Hoffman and Spaan, 1990 [classic reading]). In this model, microcirculatory vessels are compressed during systole and produce a capacitive discharge of blood that accelerates flow from the microcirculation to the coronary venous system (Fig. 49-6). At the same time, the upstream capacitive discharge impedes systolic coronary arterial inflow. Although this explains the phasic variations in coronary arterial inflow and venous outflow, as well as its transmural distribution in systole, vascular capacitance cannot explain compressive effects related to elevated tissue pressure during diastole. Thus intramyocardial capacitance, compressive changes in effective coronary backpressure, increases in systolic coronary resistance, and a time-varying driving pressure all contribute to the compressive determinants of phasic systolic coronary blood flow.
Transmural Variations in Minimum Coronary Resistance (R2) and Diastolic Driving Pressure
The subendocardial vulnerability to compressive determinants of vascular resistance is partially compensated for by a reduced minimal resistance resulting from an increased arteriolar and capillary density. Because of this vascular gradient, subendocardial flow during maximal pharmacologic vasodilation of the nonbeating heart is greater than subepicardial perfusion. Coronary vascular resistance in the maximally vasodilated heart also is pressure-dependent, reflecting passive distention of arterial resistance vessels. Thus the instantaneous vasodilated value of coronary resistance obtained at a normal coronary distending pressure will be lower than that at a reduced pressure.
The precise determinants of the effective driving pressure for diastolic perfusion continue to be controversial. Most experimental studies demonstrate that the effective back pressure to flow in the heart is higher than right atrial pressure. This has been termed zero flow pressure (Pf=0) and its minimum value is approximately 10 mm Hg in the maximally vasodilated heart. This increases to values close to LV diastolic filling pressure when preload is elevated above 20 mm Hg. Elevated preload reduces coronary driving pressure and diminishes subendocardial perfusion. It is particularly important in determining flow when coronary pressure is reduced by a stenosis, as well as in the failing heart.
Structure and Function of the Coronary Microcirculation
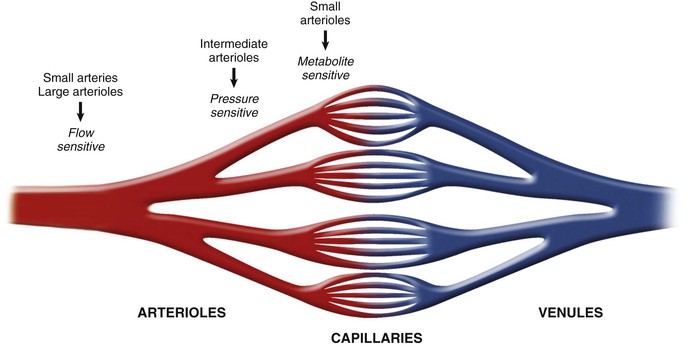
The schematics in Figures 49-4 and 49-5 suggest a fairly localized site for the control of coronary vascular resistance that is useful for conceptualizing the major determinants of coronary vascular resistance. In fact, individual coronary resistance arteries are a longitudinally distributed network and in vivo studies of the coronary microcirculation have demonstrated considerable spatial heterogeneity of specific resistance vessel control mechanisms3,4,6 (Fig. 49-7). Each resistance vessel needs to dilate in an orchestrated fashion to meet the needs of the downstream vascular bed, which is frequently removed from the site of metabolic control of coronary resistance. This can be accomplished independently of metabolic signals by sensing physical forces such as intraluminal flow (shear stress–mediated control) or intraluminal pressure changes (myogenic control). Epicardial arteries (more than 400 µm in diameter) serve a conduit artery function, with diameter primarily regulated by shear stress, and contribute little pressure drop (less than 5%) over a wide range of coronary flow. Coronary arterial resistance vessels can be divided into small arteries (100 to 400 µm), which regulate their tone in response to local shear stress and luminal pressure changes (myogenic response), and arterioles (<100 µm), which are sensitive to changes in local tissue metabolism and directly control perfusion of the low-resistance coronary capillary bed (Fig. 49-8; see also Fig. e49-2 ).3,4 Capillary density of the myocardium averages 3500/mm2 (resulting in an average intercapillary distance of 17 µm), which is greater in the subendocardium than in the subepicardium.
).3,4 Capillary density of the myocardium averages 3500/mm2 (resulting in an average intercapillary distance of 17 µm), which is greater in the subendocardium than in the subepicardium.
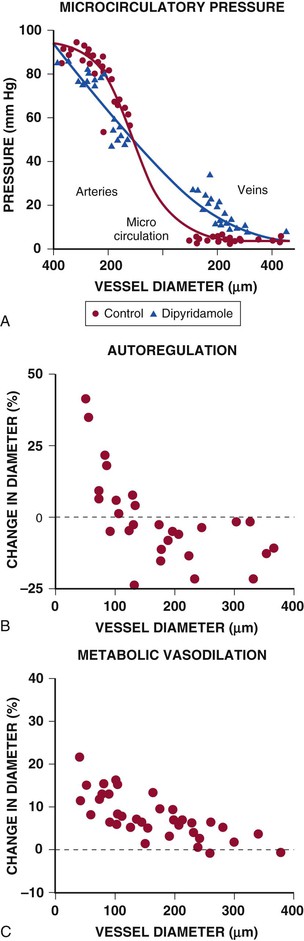
Under resting conditions, most of the pressure drop in the microcirculation arises in resistance arteries between 50 and 200 µm, with little pressure drop occurring across capillaries and venules at normal flow levels (Fig. e49-2A).4 After pharmacologic vasodilation with dipyridamole, resistance artery vasodilation attenuates the precapillary pressure drop in arterial resistance vessels. At the same time, there is an increased pressure drop and redistribution of resistance to venular vessels, in which smooth muscle relaxation is limited and the already low resistance is fairly fixed.
Considerable heterogeneity in microcirculatory vasodilation is evident during physiologic adjustments in flow. For example, as pressure is reduced during autoregulation, dilation is accomplished primarily by arterioles smaller than 100 µm, whereas larger resistance arteries tend to constrict because of the reduction in perfusion pressure (Fig. e49-2B).4 By contrast, metabolic vasodilation results from a more uniform vasodilation of resistance vessels of all sizes (Fig. e49-2C).4 Similar inhomogeneity in resistance vessel dilation occurs in response to endothelium-dependent agonists as well as pharmacologic vasodilators.
A unique component of subendocardial coronary resistance vessels is the transmural penetrating arteries that course from the epicardium to the subendocardial plexus.2 These vessels not only are less sensitive to metabolic signals but are also removed from the metabolic stimuli that develop when ischemia is confined to the subendocardium. As a result, local control by altered shear stress and myogenic relaxation to local pressure become very critical determinants of diameter in this “upstream” resistance segment. Even during maximal vasodilation, this segment creates an additional longitudinal component of coronary vascular resistance that must be traversed before the arteriolar microcirculation is reached. Because of this greater longitudinal pressure drop, the microcirculatory pressures in subendocardial coronary arterioles are lower than in the subepicardial arterioles.4
Intraluminal Physical Forces Regulating Coronary Resistance.
Because much of the coronary resistance vasculature can be upstream from the effects of metabolic mediators of control, local vascular control mechanisms are critically important in orchestrating adequate regional tissue perfusion to the distal microcirculation. The differential expression of mechanisms that is evident among different sizes and classes of coronary resistance vessels coincides with their function.
Myogenic Regulation.
The myogenic response refers to the ability of vascular smooth muscle to oppose changes in coronary arterial diameter.3 Thus vessels relax when distending pressure is decreased and constrict when distending pressure is elevated (Fig. 49-9A). Myogenic tone is a property of vascular smooth muscle and occurs across a large size range of coronary resistance arteries in animals as well as in humans. Although the cellular mechanism is uncertain, it is dependent on vascular smooth muscle calcium entry, perhaps through stretch-activated L-type Ca2+ channels, eliciting cross-bridge activation. The resistance changes arising from the myogenic response tend to bring local coronary flow back to the original level. Myogenic regulation has been postulated to be one of the important mechanisms of the coronary autoregulatory response and, in vivo, appears to primarily occur in arterioles smaller than 100 µm (e.g., during autoregulation); (see Fig. e49-2B).
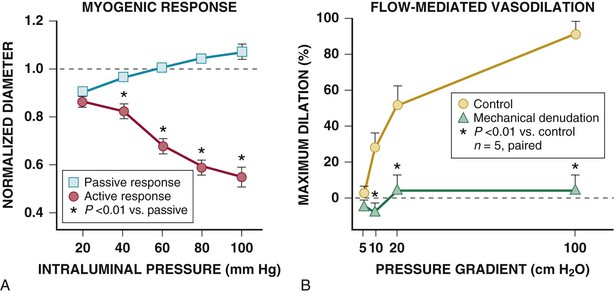
Flow-Mediated Resistance Artery Control.
Coronary small arteries and arterioles also regulate their diameter in response to changes in local shear stress (see Fig. 49-9B). Flow-induced dilation in isolated coronary arterioles is endothelium-dependent and mediated by NO, because it could be abolished with an l-arginine analogue. By contrast, isolated atrial vessels from patients undergoing cardiac surgery exhibit flow-mediated vasodilation that is mediated by EDHF.7,8 The disparity with animal studies may reflect age or species variability in the relative importance of EDHF versus NO in the coronary circulation. The mechanisms also appear to vary as a function of vessel size, with studies in pigs demonstrating that hyperpolarization regulates epicardial conduit arteries,9 and NO predominates in the resistance vasculature. Finally, EDHF may represent a compensatory pathway that normally is inhibited by NO and becomes upregulated in acquired disease states in which NO-mediated vasodilation is impaired.7 More recent studies have demonstrated that this factor appears to be hydrogen peroxide.10 Despite the variability in isolated vessels, blocking NOS with an l-arginine analogue in the coronary circulation of humans reduces vasodilation to pharmacologic endothelium-dependent agonists and attenuates flow increases during metabolic vasodilation. This demonstrates that NO-mediated vasodilation plays a role in determining physiologic vascular tone in some segments of the coronary resistance vasculature.
Metabolic Mediators of Coronary Resistance Vessel Control.
Although increasing knowledge has emerged regarding the distribution of coronary microvascular resistance, there is still no consensus regarding specific mediators of metabolic vasodilation.11 Coronary resistance in any segment of the microcirculation represents the integration of local physical factors (e.g., pressure and flow), vasodilator metabolites (e.g., adenosine, Po2, and pH), autacoids, and neural modulation. Each of these mechanisms contributes to net coronary vascular smooth muscle tone, which may ultimately be controlled by opening and closing vascular smooth muscle adenosine triphosphate (ATP)-sensitive K+ (KATP) channels. There is considerable redundancy in the available local control mechanisms.2 Because of this, blocking single mechanisms fails to alter coronary autoregulation or metabolic flow regulation at normal coronary pressures. This redundancy can, however, be unmasked by stressing the heart and evaluating flow regulation at reduced pressures distal to a coronary stenosis at rest or during exercise.2 Some of the candidates proposed and their role in metabolic resistance control and ischemia-induced vasodilation are summarized here; for more extensive discussion, see the classic reference by Feigl (1983).
Adenosine.
There has been a longstanding interest in the role of adenosine as a metabolic mediator of resistance artery control. It is released from cardiac myocytes when the rate of ATP hydrolysis exceeds its synthesis during ischemia. Its production and release also increase with myocardial metabolism. Adenosine has an extremely short half-life (less than 10 seconds) caused by its rapid inactivation by adenosine deaminase. It binds to A2 receptors on vascular smooth muscle, increases cyclic adenosine monophosphate (cAMP), and opens KATP6 and intermediate calcium-activated potassium channels.12 Adenosine has a differential effect on coronary resistance arteries, primarily dilating vessels smaller than 100 µm.4 Although adenosine has no direct effect on larger resistance arteries and conduit arteries, these dilate through endothelium-dependent vasodilation from the concomitant increases in local shear stress as arteriolar resistance falls.3 Despite the attractiveness of adenosine as a local metabolic control mechanism, there is now substantial in vivo experimental data to demonstrate convincingly that it is not required for adjusting coronary flow to increases in metabolism or autoregulation.6 It may, however, contribute to vasodilation during hypoxia as well as during acute exercise-induced myocardial ischemia distal to a stenosis.2
ATP-Sensitive K+ Channels.
Coronary vascular smooth muscle KATP channels are tonically active, contributing to coronary vascular tone under resting conditions. Preventing KATP channel opening with glibenclamide causes constriction of arterioles smaller than 100 µm, reduces coronary flow, and accentuates myocardial ischemia distal to a coronary stenosis by overcoming intrinsic vasodilator mechanisms.2 The KATP channels can modulate both the coronary metabolic and autoregulatory responses. It is a potentially attractive mechanism, because many of the other candidates for metabolic flow regulation (e.g., adenosine, NO, beta2-adrenoreceptors, prostacyclin) are ultimately affected by blocking this pathway. It is likely that KATP channel opening is a common effector rather than sensor of metabolic activity or of autoregulatory adjustments in flow. It is also possible that the reductions in coronary flow observed after blocking KATP channel vasodilation are pharmacologic, caused by vasoconstriction of the microcirculation that overcomes intrinsic vasodilator stimuli, as seen when other potent vasoconstrictors (e.g., endothelin or vasopressin) are administered at pharmacologic doses.
Oxygen Sensing.
Although a potent coronary vasodilator stimulus, the role of local Po2 in the regulation of arteriolar tone remains unresolved. Coronary flow increases in proportion to reductions in arterial oxygen content (reduced Po2 or anemia) and there is a twofold increase in perfused capillary density in response to hypoxia. The underlying mechanism may involve the release of NO and ATP (which stimulates vascular endothelial P2 receptors to produce NO) from red blood cells, when intravascular Po2 levels drop.1 Studies demonstrating a direct effect of oxygen on metabolic or autoregulatory adjustments are lacking, however, and the vasodilator response to reduced arterial oxygen delivery may simply reflect the close coupling between myocardial metabolism and flow.
Acidosis.
Arterial hypercapnia and acidosis (Pco2) are potent stimuli that have been demonstrated to produce coronary vasodilation independent of hypoxia. Whereas their precise role in the local regulation of myocardial perfusion remains unclear,1 it seems reasonable that some of the vasodilation occurring with increased myocardial metabolism could arise from increased myocardial CO2 production and tissue acidosis in the setting of acute ischemia.
Neural Control of Coronary Conduit and Resistance Arteries
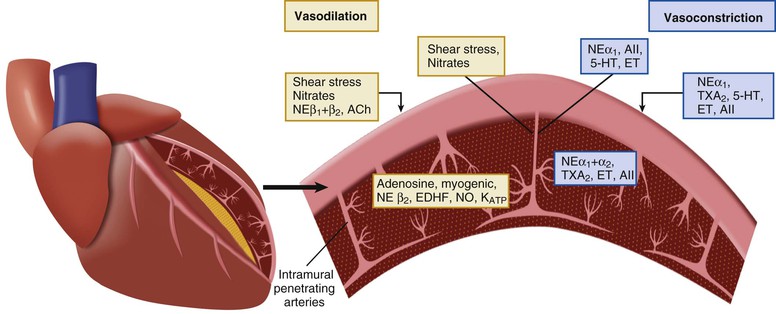
Sympathetic and vagal nerves innervate coronary conduit arteries and segments of the resistance vasculature. Neural stimulation affects tone through mechanisms that alter vascular smooth muscle as well as by stimulating the release of NO from the endothelium. Diametrically opposite effects can occur in the presence of risk factors that impair endothelium-dependent vasodilation. Their actions in normal and pathophysiologic states are summarized in Table 49-1.
Cholinergic Innervation
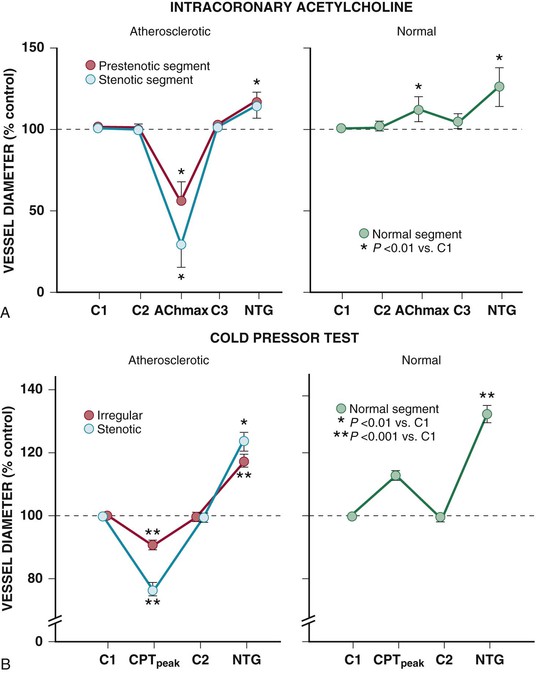
Resistance arteries dilate to acetylcholine, resulting in increases in coronary flow. In conduit arteries, acetylcholine normally causes mild coronary vasodilation. This reflects the net action of a direct muscarinic constriction of vascular smooth muscle counterbalanced by an endothelium-dependent vasodilation caused by direct stimulation of NOS and an increased flow-mediated dilation from concomitant resistance vessel vasodilation. The response in humans with atherosclerosis or risk factors for CAD is distinctly different. The resistance vessel dilation to acetylcholine is attenuated and the reduction in flow-mediated NO production leads to net epicardial conduit artery vasoconstriction, which is particularly prominent in stenotic segments (Fig. 49-10A).
Sympathetic Innervation
Under basal conditions, there is no resting sympathetic tone in the heart and thus there is no effect of denervation on resting perfusion. During sympathetic activation, coronary tone is modulated by norepinephrine released from myocardial sympathetic nerves, as well as by circulating norepinephrine and epinephrine.13 In conduit arteries, sympathetic stimulation leads to alpha1 constriction as well as beta-mediated vasodilation. The net effect is to dilate epicardial coronary arteries. This dilation is potentiated by concomitant flow-mediated vasodilation from metabolic vasodilation of coronary resistance vessels. When NO-mediated vasodilation is impaired, alpha1 constriction predominates and can dynamically increase stenosis severity in asymmetrical lesions where the stenosis is compliant. This is one of the mechanisms by which ischemia can be provoked during cold pressor testing (Fig. 49-10B).
The effects of sympathetic activation on myocardial perfusion and coronary resistance vessel tone are complex and dependent on the net actions of beta1-mediated increases in myocardial oxygen consumption (resulting from increases in the determinants of myocardial oxygen consumption), direct beta2-mediated coronary vasodilation, and alpha1-mediated coronary constriction. Under normal conditions, exercise-induced beta2-adrenergic “feed-forward” dilation predominates, resulting in a higher flow relative to the level of myocardial oxygen consumption.6 This neural control mechanism produces transient vasodilation before the buildup of local metabolites during exercise and prevents the development of subendocardial ischemia during abrupt changes in demand. After nonselective beta blockade, sympathetic activation unmasks alpha1-mediated coronary artery constriction. Although flow is mildly decreased, oxygen delivery is maintained by increased oxygen extraction and a reduction in coronary venous Po2 at similar levels of cardiac workload. Intense alpha1-adrenergic constriction can overcome intrinsic stimuli for metabolic vasodilation to result in ischemia in the presence of pharmacologic vasodilator reserve.13 The role of pre- and postsynaptic alpha2 responses is controversial. They appear to have a less significant role in controlling flow. This partly reflects the competing effects of presynaptic alpha2 receptor stimulation, leading to reduced vasoconstriction by inhibiting norepinephrine release.
Paracrine Vasoactive Mediators and Coronary Vasospasm
There are a large number of paracrine factors that can affect coronary tone in normal and pathophysiologic states that are unrelated to normal coronary circulatory control. The most important of these are summarized in Table 49-1 and Figure 49-7 (see also Fig. e49-1). Paracrine factors are released from epicardial artery thrombi after activation of the thrombotic cascade initiated by plaque rupture. They can modulate epicardial tone in regions near eccentric ulcerated plaques that are still responsive to stimuli that alter smooth muscle relaxation and constriction, leading to dynamic changes in the physiologic significance of a stenosis. Paracrine mediators also can have differential effects on downstream vessel vasomotion that are dependent on vessel size (conduit arteries versus resistance arteries) as well as on the presence of a functionally normal endothelium, because many also stimulate the release of NO and EDHF.
Serotonin released from activated platelets causes vasoconstriction in normal and atherosclerotic conduit arteries and can increase the functional severity of a dynamic coronary stenosis through superimposed vasospasm. By contrast, it dilates coronary arterioles and increases coronary flow through the endothelium-dependent release of NO. In atherosclerosis or circumstances in which NO production is impaired, the direct effects on smooth muscle predominate and the response of the microcirculation is converted to vasoconstriction. As a result, serotonin release generally exacerbates ischemia in CAD.
Thromboxane A2 is a potent vasoconstrictor that is a product of endoperoxide metabolism and released during platelet aggregation. It produces vasoconstriction of conduit arteries as well as isolated coronary resistance vessels and can accentuate acute myocardial ischemia.
Adenosine diphosphate (ADP) is another platelet-derived vasodilator that relaxes coronary microvessels as well as conduit arteries. It is mediated by NO and abolished by removing the endothelium.
Thrombin normally leads to vasodilation in vitro that is endothelium-dependent and mediated by the release of prostacyclin as well as NO. In vivo, it also releases thromboxane A2, leading to vasoconstriction in epicardial stenoses in which endothelium-dependent vasodilation is impaired. In the coronary resistance vasculature, it acts as an endothelium-dependent vasodilator and increases coronary flow.
Coronary Vasospasm
Coronary spasm results in transient functional occlusion of a coronary artery that is reversible with nitrate vasodilation. It most commonly occurs in the setting of a coronary stenosis, leading to dynamic stenosis behavior that can dissociate the effects on perfusion from anatomic stenosis severity (see Chapter 20). In CAD, endothelial disruption probably plays a role in focal vasospasm. In this setting, the normal vasodilation from autacoids and sympathetic stimulation is converted into a vasoconstrictor response because of the lack of competing endothelium-dependent vasodilation. Nevertheless, although impaired endothelium-dependent vasodilation is a permissive factor for vasospasm, it is not causal, and a trigger is required (e.g., thrombus formation or sympathetic activation).
The mechanisms responsible for variant angina with normal coronary arteries, or Prinzmetal angina, are less clear. Data from animal models have pointed to sensitization of intrinsic vasoconstrictor mechanisms.14 Coronary arteries demonstrate supersensitivity to vasoconstrictor agonists in vivo and in vitro as well as reduced vasodilator responses. Some studies have demonstrated that Rho, a guanosine triphosphate (GTP)-binding protein, can sensitize vascular smooth muscle to calcium by inhibiting myosin phosphatase activity through the effector protein Rho kinase.
Pharmacologic Vasodilation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


