2 Coronary Atherosclerosis
Etiology and Pathogenesis
The concept that endothelial injury is an inciting event in atherosclerosis is common to most theories of pathogenesis. Endothelial injury is a component of the earliest stages of atherosclerosis, the formation of lesions that can be detected only at autopsy, the fatty streak (Fig. 2-1A). Most of the well-characterized risk factors for atherosclerosis (hypertension, diabetes mellitus, cigarette smoking, hyperlipidemia, advanced age, elevated plasma homocysteine concentrations) injure the endothelium, initiating a chain of events, all attributes of atherosclerosis: smooth muscle cell (SMC) proliferation, inflammatory cell recruitment, and lipid deposition within the blood vessel (Fig. 2-1B). Though still early in development, potential diagnostic and/or therapeutic approaches based upon inflammatory signaling pathways now known to be important in atherogenesis hold promise.
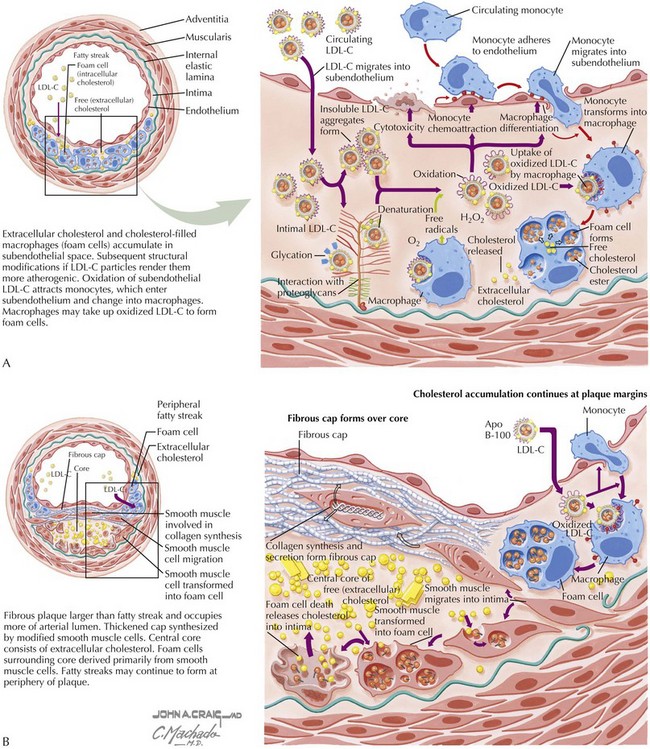
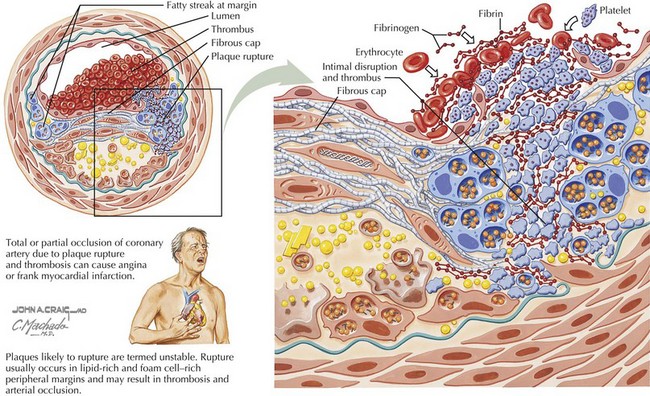
Endothelial injury and the subsequent events that occur in the vessel wall initiate the progression from stable to unstable atherosclerotic plaques, ultimately leading to the rupture of unstable plaques, thrombosis of the vessel, and, in many cases, MI (Fig. 2-2). Lesion development in the medium and small vessels of cerebral vessels leads to stroke, and in renal and mesenteric vasculature contributes to diabetic complications.
An abundance of evidence suggests that atherosclerotic lesions, at least in part, result from an excessive inflammatory response. For example, although elevated low-density lipoprotein cholesterol (LDL-C) is a risk factor for premature atherosclerosis, the LDL-C must undergo oxidative modification to cause damage to the arterial wall. Cytokines, growth factors, and oxidative stress may also contribute to atherosclerosis by mechanisms that are independent of LDL-C oxidation. Any of these mediators can rapidly react with and inactivate nitric oxide, enhancing proatherogenic mechanisms such as leukocyte adhesion to endothelium, impaired vasorelaxation, and platelet aggregation (Fig. 2-3).
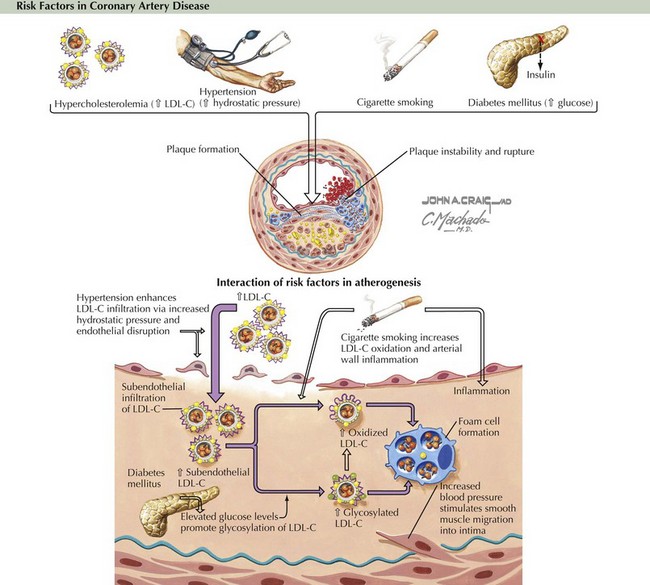
Figure 2-3 Classical risk factors in coronary artery disease: Relationship to excessive inflammatory responses.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


