dysfunction of the endothelial lining of the vessel
inflammation of the vascular wall
build up of lipids, cholesterol, and inflammatory cells in the vessel wall
accumulation of cellular debris within the intima and subintimal layers of the vessel.
tobacco toxins
oxidized low-density lipoprotein (LDL)
advanced glycation end-products
elevated homocysteine
infectious agents.
reduced bioavailability of endothelial nitric oxide (NO), an important vasodilator, anti-thrombotic, and antiproliferative agent
increased generation of potent vasoconstrictor agents endothelin-1 and angiotensin-II, promoting cell migration and growth
dysfunctional endothelial cells express adhesion molecules and secrete chemokines, promoting cell migration and adhesion
local thrombotic balance is altered as levels of plasminogen activator inhibitor (PAI) and tissue factor are increased
tissue plasminogen activator (t-PA) and thrombomodulin are reduced
low NO release results in increased platelet activation and adhesion.
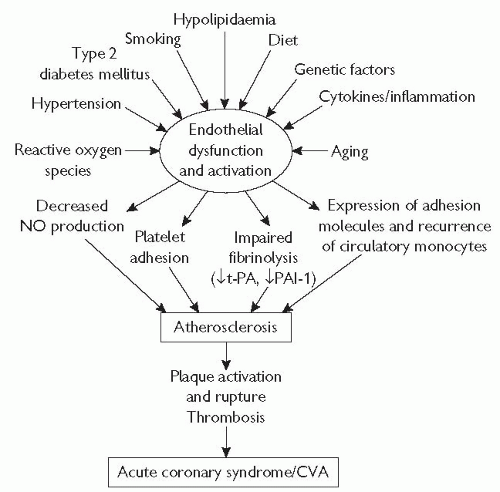 Fig. 5.1 Endothelial dysfunction is the underlying process in atherosclerosis, from lesion initiation and progression, through to acute cardiovascular events. Endothelial dysfunction is caused by a variety of genetic factors (and aging), as well as environmental factors that can be modified. CVA = cerebrovascular accident. |
Circulating leucocytes, predominantly monocytes, are attracted and bind to activated endothelial cells, followed by migration into the subendothelial layer, where they transform into macrophages.
They act as local ‘scavenger’ cells with the capacity to take up modified LDL cholesterol (LDL-C), ultimately becoming the characteristic ‘foam cells’ of established atherosclerosis.
The earliest lesions are known as ‘fatty streaks’, which consist predominantly of lipid-accumulating macrophages and foam cells.
These lesions may develop into fibrous plaques, as a consequence of further lipid accumulation accompanied by local migration, proliferation, and fibrous transformation of smooth muscle cells.
These cells are responsible for the deposition of extracellular connective tissue matrix, leading to formation of a fibrous cap, which overlies a central core, consisting of foam cells, extracellular lipid, necrotic cellular debris, and a mixture of other inflammatory cells including T-lymphocytes.
This process is facilitated by ongoing endothelial dysfunction, together with local generation of powerful mitogens such as platelet-derived growth factor (PDGF), transforming growth factor-beta (TGF-β), and insulin-like growth factor (IGF) from endothelial cells, macrophages, and activated platelets.
Further growth of the plaque initially causes outward remodelling of the vessel wall, minimizing the impact on the cross-sectional area of the lumen and the vessel’s ability to deliver blood.
Progressive plaque accumulation results in luminal narrowing, and ultimately vessel obstruction.
Blood flow through arteries causes local generation of fluid ‘shearstress’, which influences the biology of the underlying endothelial cells.
High laminar shear (from blood flowing quickly through a straight vessel) favours the generation of NO, which helps maintain the functional integrity of the vessel.
In contrast, low shear, or ‘differential’ shear caused by turbulent flow, causes dysfunction of the underlying endothelial cells, which facilitates initiation and progression of atheroma.
This helps explain why plaques are found more commonly at sites of vessel branching or curvature, which experience more dramatic and abrupt changes and irregularities of direction and velocity of blood flow.
These shear-stress-mediated effects are most marked in vessels that carry a high (basal) blood flow, such as the coronary, carotid, renal, and ilio-femoral arteries, in which the majority of clinically important atherosclerotic lesions develop.
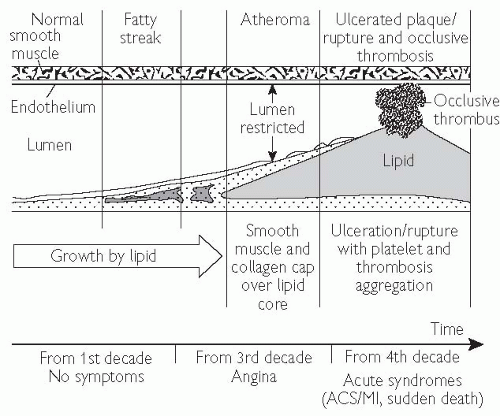 Fig. 5.2 Cellular interactions in the development and progression of atherosclerosis. VSMC, vascular smooth muscle cells. ACS = acute coronary syndrome; MI = myocardial infarction. Reproduced with permission from Weissberg PL (2003). Atherogenesis: current understanding of the causes of atheroma. Heart 83: 247-52. |
 Risk factors for coronary artery disease, p. 220), which together account for the majority of the CAD burden in the UK. Although identification and treatment of individuals with these risk factors has the potential to reduce significantly the burden of CAD, the incidence of and death rate from MI remain high.
Risk factors for coronary artery disease, p. 220), which together account for the majority of the CAD burden in the UK. Although identification and treatment of individuals with these risk factors has the potential to reduce significantly the burden of CAD, the incidence of and death rate from MI remain high.
Collagen, tissue factor, and other factors activate platelets and trigger the coagulation cascade.
This leads to acute thrombosis, which may rapidly occlude the vessel.
This results in MI of this vascular territory, usually characterized by ST-segment elevation on the electrocardiogram (ECG).
Coronary thrombosis is a dynamic process in vivo, and may be reversed, at least in part, by activation of t-PA and proteins C and S of the intrinsic antithrombotic/fibrinolytic system.
Acute, subtotal occlusion of the vessel typically causes acute symptomatic deterioration and non-ST-segment elevation MI (NSTEMI) or unstable angina.
Atheromatous plaques that have a thin fibrous cap and a large necrotic lipid core, and contain a high proportion of inflammatory cells and mediators, and are particularly predisposed to destabilization or rupture, with consequent thrombosis.
Conversely, plaques with a smaller lipid pool, thicker fibrous caps, and less inflammatory activity are more stable and less prone to rupture.
Several studies have shown that well over half of all MIs are caused by acute destabilization of plaques that were previously not obstructing flow in the vessel, suggesting that the likelihood of an acute coronary event is more closely related to the stability of the plaque rather than the severity of the stenosis.
the Framingham risk-scoring system
the Sheffield tables
the European SCORE charts.
young subjects with multiple risk factors
subjects with a family history of premature cardiovascular disease
certain ethnic groups including individuals of South Asian racial origin living in the UK.
C-reactive protein (CRP) levels
ultrasound assessment of carotid arterial intima-media thickness
detection of coronary artery calcification with computed tomography (CT) scanning.
Calculation of risk is unnecessary in subjects who already have clinical atherosclerotic disease, as secondary prevention measures are mandatory in these individuals.
Similarly, risk calculation is not required for subjects with diabetes mellitus, whose risk approaches that of subjects with established atherosclerosis.
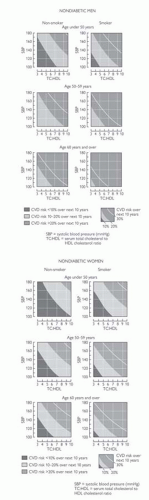 Fig. 5.3 Reproduced with permission from 2004 Joint British Societies Risk Prediction Tables. © The University of Manchester. CVD = cardiovascular disease. |
between 1971 and 2002, the percentage of older people (aged 65 years and over) in the UK, increased from 13% to 16%, and is projected to rise to 23% in the next 25 years
aging is a major risk factor for atherosclerotic disease, due to the degenerative process associated with aging per se, together with the cumulative impact of the worsening risk-factor profile that develops with increasing age
by the age of 70 years, 15% of men and 9% of women have symptomatic CAD, increasing to 20% by the age of 80 years
Over 40 000 premature (<75 years) deaths are caused by CAD, 22% of premature deaths in men and 13% of those in women
CAD is more common in men than women and the onset tends to be earlier in men.
The incidence of coronary heart disease (CHD) in women increases rapidly at menopause, and is similar to that seen in men in the population over 65 years.
Although less common, the disease remains one of the biggest killers of women; for example, the age-adjusted mortality rates from heart disease are four to six times higher than their mortality rates from breast cancer.
Female sex hormones probably contribute to the lower risk of atherosclerotic disease in premenopausal women.
The risk of ischaemic heart disease is reduced by up to 40% in women using HRT. HRT users are typically healthier than non-users, suggesting that these results could be explained by selection bias.
Several large, randomized controlled trials of HRT in postmenopausal women (WHI (Women’s Health Initiative), HERS (Heart and Estrogen/
Progestin Study)/HERS II, ESPRIT (Estrogen in the Prevention of Reinfarction Trial), ERA (Estrogen Replacement and Atherosclerosis)) with and without atherosclerosis have now clearly shown that HRT does not reduce the risk of cardiovascular events.
HRT may, in fact, even lead to a small but statistically significant increase in morbidity and mortality from cardiovascular and other diseases including gynaecologic malignancy.
HRT should not be recommended for primary or secondary prevention of atherosclerotic disease in postmenopausal women.
CAD is a multifactorial, polygenic disorder, caused by interactions between lifestyle, the environment, and the effects of variations in the genetic sequence of a number of genes.
The family history is considered to be significant when atherosclerotic disease presents in a first-degree male relative before the age of 55 years, or before 65 years in a female relative.
A positive family history is associated with a 75% increase in risk in men, and an 84% increase in women. The risk is more than doubled if both parents are affected.
Risk factors for coronary artery disease | ||||
|---|---|---|---|---|
|
Inflammation
Fibrinogen, and other factors involved in thromboregulation
Homocysteine
Oxidative stress
Asymmetric dimethylarginine
Age-standardized mortality from CAD is around 50% higher in individuals of South Asian racial origin living in the UK compared to white individuals.
Although an increased prevalence of risk factors explains much of this risk (high triglycerides, low high-density lipoprotein (HDL), insulin resistance, and reduced physical activity), genetic factors are thought to contribute significantly.
The observed incidence of CAD is lower in black individuals of West Indian and African origin in the UK, although the incidence of stroke is greater than in the Caucasian population.
The burden of risk attributable to psychological stress is more difficult to quantify.
Increased work stress, lack of social support, hostile personality type, anxiety, and depression are most consistently associated with increased atherosclerosis risk.
Smoking increases the risk of CAD by approximately 50%, with mortality from any cardiovascular disease around 60% higher in smokers (and 85% higher in heavy smokers) compared to non-smokers.
In the UK today, around 13 million adults (28% of men and 26% of women) smoke cigarettes.
Although the number of smokers has declined substantially over the past 50 years, this trend has slowed down in the young, and the number of teenage girls that smoke has recently increased.
Over 30 000 cardiovascular deaths per year (14% in men and 12% in women, with an even higher proportion of premature deaths) are attributable to smoking.
Second-hand smoke (smoke that has been exhaled by a smoker) can also increase the risk of CAD by around 25%.
Stopping smoking carries almost immediate benefit, and although the long-term benefits are greatest in those who stop smoking before the age of 40 years, stopping in middle age is also beneficial. For example, in those aged 30-59 years who stop smoking after a MI, the 5-year mortality is 10%, compared with 14% in those who continue to smoke.
Individuals at increased risk of atherosclerosis should be advised to stop smoking. See Figs. 5.4. and 5.5.
Evidence now exists that focused psychosocial support, nicotine replacement, and pharmacological therapy are effective in helping individuals stop smoking. These services are delivered in an integrated fashion by smoking-cessation clinics, which can deliver a fourfold increase in the likelihood that a smoker will quit, vs. use of willpower alone.
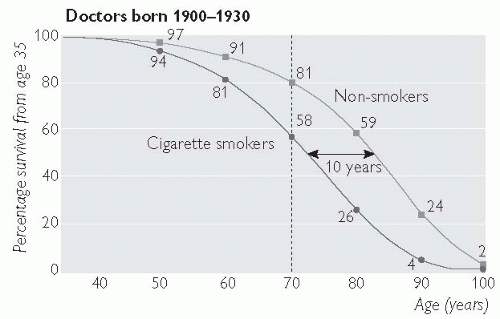 Fig. 5.4 Survival from age 35 years for continuing cigarette smokers and lifelong non-smokers among UK male doctors born 1900-1930, with percentages alive at each decade of age (adapted and reproduced with permission from Doll R, Peto R, Boreham J, Sutherland I (2004). Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ 328(7455): 1519). |
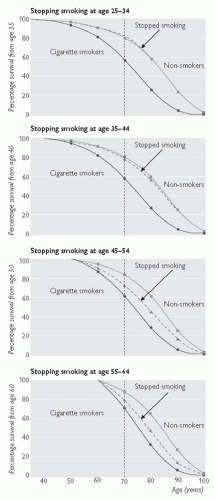 Fig. 5.5 Effects on survival of stopping smoking cigarettes at age 25-34 years (effect from age 35 years), age 35-44 years (effect from age 40 years), age 45-54 years (effect from age 50 years), and age 55-64 years (effect from age 60 years) (adapted and reproduced with permission from Doll R, Peto R, Boreham J, Sutherland I (2004). Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ 328(7455): 1519). |
Obesity increases the risk of CAD: 25-49% of CAD in developed countries is attributable to increased body mass index (BMI).
The prevalence of obesity is increasing rapidly worldwide. In the UK, adult obesity has increased by over 50% in less than 10 years.
Of particular concern is the dramatic increase in the prevalence of obesity in children, which has almost doubled in the UK in less than 10 years. This trend is likely to exacerbate the problem in adulthood, and undo many of the other recent improvements in cardiovascular health.
Central obesity, in which excess fat is concentrated mainly in the abdomen, can be identified by a high waist-to-hip ratio, and confers a particularly high relative risk of CAD if the waist circumference is >102 cm (40 in.) in men and >88 cm (35 in.) in women.
Associated metabolic abnormalities of central obesity include high triglycerides, low HDL, high blood pressure, low-grade systemic inflammation, insulin resistance, and type 2 diabetes mellitus.
Overweight and obese individuals also tend to be less physically active and eat lower-quality diets, which contributes further to their atherogenic risk.
Diet and exercise should be considered as the first line of intervention in these individuals, together with careful surveillance for and aggressive treatment of diabetes, high blood pressure, and dyslipidaemia when present.
Atherosclerosis involves an ongoing inflammatory process from the time of lesion initiation, through progression and at the time of acute thrombotic complication.
This links risk factors and the disease mechanism in a way that is directly applicable to human patients.
Large studies have shown that low-grade elevation in markers of inflammation, most notably CRP, predicts outcomes from atherosclerotic disease and may add to prognostic information provided by traditional risk factors.
A CRP level below 1 mg/L is associated with low risk, 1-3 mg/L reflects intermediate risk, and a level above 3 mg/L is associated with a high long-term risk of vascular events.
Certain treatments that reduce coronary risk also limit inflammation, such as statins and aspirin, which may contribute to their clinical benefits.
The incremental value of CRP as a marker of risk and target of therapy in global risk management is currently a controversial topic that requires clarification by prospective randomized trials.
The genetic disease homocystinuria is associated with aggressive, premature atherosclerosis.
Strong epidemiologic and mechanistic evidence suggests that even modest increases in homocysteine levels increase the progression of atherosclerosis.
Vascular inflammation and oxidative stress appear to be the responsible mechanisms.
Dietary supplementation with folic acid, alone or in combination with vitamins B6 and B12, can reduce levels of homocysteine and improve aspects of vascular biology in vivo as well as in vitro.
Until results of large, outcome-driven, randomized clinical trials are available, folate ± B6/B12 supplementation cannot be routinely recommended for prevention of atherosclerotic disease.
total fat intake should be reduced to below 30% of total calorie intake
intake of saturated fat and foods high in ‘trans’ fatty acids should be limited, and replaced with monounsaturated fat (canola and olive oil)
dietary salt intake should be reduced
intake of fresh fruits and vegetables should be increased (≥5 portions per day)
fish consumption, especially oily fish, should be encouraged, with evidence suggesting that at least one fish meal, and ideally 2-3 per week can reduce the incidence of heart attack and stroke.
The contribution of physical inactivity to CAD deaths is difficult to quantify; however, people who are physically active appear to have a lower risk of CAD.
This is mediated, at least in part by weight loss, a reduction in blood pressure, and improvement of the lipid profile (particularly increased HDL).
Regular, aerobic exercise of moderate intensity should be undertaken ≥3 times per week for at least 30 minutes, but greater frequency and duration of exercise is associated with increasing benefits.
The patient often has a headache and occasionally visual disturbance.
Proteinuria and haematuria are often present.
This is a medical emergency requiring immediate treatment to prevent rapid progression to renal failure, heart failure, and/or stroke.
Untreated, the 1-year mortality is approximately 90%.
fasting glucose
urea and electrolytes (U&E)
urinalysis for blood and protein
Table 5.1 Blood pressure (BP) classification | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||

Diabetic nephropathy, renovascular disease, glomerulonephritis, vasculitides, chronic pyelonephritis, polycystic kidneys
Conn’s and Cushing’s syndromes, glucocorticoid-remediable hypertension, phaeochromocytoma, acromegaly, hyperparathyroidism
Aortic coarctation, pregnancy-induced hypertension and preeclampsia, obesity, excessive dietary salt or licquorice intake, drugs (non-steroidal anti-inflammatory drugs (NSAIDs), sympathomimetics, illicit stimulants, e.g. amphetamine, MDMA (‘ecstasy’), and cocaine
Tortuous arteries, with thickened bright walls (‘silver wiring’)
Arteriovenous nipping (narrowing in a vein where crossed by an artery)
Flame haemorrhages and cotton wool spots (small retinal bleeds and exudates)
Papilloedema
Patients with malignant hypertension or with persistent BP>160/100 mmHg after lifestyle measures
Subjects with BP>140/90 mmHg after lifestyle measures who have evidence of end-organ damage (left ventricular hypertrophy (LVH) on ECG, proteinuria or retinopathy) or a calculated 10-year risk of a cardiovascular event ≥20%
Minimize dietary salt intake (<100 mmol/day)
Reduce alcohol to <21 units (men) and <14 units (women) per week
Take regular aerobic exercise if not contraindicated (at least 30 min, 3x/week)
Achieve and maintain healthy BMI (20-25 kg/m2)
Consume at least 5 portions/day of fresh fruit and vegetables
Stop smoking, and reduce dietary fat content, especially saturated and trans-fatty acids.
Thiazide diuretics are effective and cheap and widely recommended as first-line antihypertensive therapy.
Although calcium-channel blockers may be less protective than other agents against the development of heart failure, their safety and effectiveness in preventing atherosclerotic events is now confirmed, despite previous concerns.
Table 5.2 Indications for antihypertensive drugs | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||

This is based on the A-B-C-D principle (A = ACE-I or ARB, B = β-blocker, C = calcium-channel antagonist, D = diuretic).
A or B are effective first-line drugs in the young, who typically have high-renin hypertension that responds well to these drug classes.
C or D are more effective first-line agents in the elderly and black individuals, who typically have lower levels of renin and are less responsive to these agents.
Drugs can be substituted or added in a stepwise fashion according to the response, until BP is controlled.
The lipid-lowering arm of the ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial) study was terminated early due to a sizeable reduction in major vascular events seen in patients with hypertension and ‘average’ cholesterol levels treated with atorvastatin 10 mg daily for less than 4 years.
Aspirin 75 mg daily is recommended in hypertensive patients with evidence of clinical atherosclerotic disease or ≥20% 10-year cardiovascular event risk, after adequate BP control is achieved (<150/90 mmHg).
Statin therapy should be initiated in these high-risk individuals, regardless of baseline cholesterol levels.
Table 5.3 Combining antihypertensive drugs: (The BHS A-B-C-D principle) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||

is over 50 years of age
has clinical atherosclerotic disease
has ≥other risk factors for atherosclerosis
has clinical signs of hyperlipidaemia (xanthomata, xanthelasmata, or corneal arcus at age <50 years)
has a family history of premature CAD or hyperlipidaemia.
Normal ranges for plasma lipid levels | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 5.4 Primary hyperlipidaemias | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
increase clearance of circulating LDL by upregulating LDL-receptor expression.
are extremely effective at lowering cholesterol levels; for example, the most potent statins, atorvastatin and rosuvastatin can more than halve LDL-C levels at higher doses.
increase HDL and decrease triglyceride levels.
Data from large randomized clinical trials have shown a consistent reduction (around 25-30%) in the relative risk of major cardiovascular events with the use of atorvastatin, pravastatin, and simvastatin in both the primary (ASCOT, WOSCOPS (West of Scotalnd Coronary Prevention Study), HPS, CARDS (Collaborative Atorvastatin Diabetes Study)) and secondary prevention settings (MIRACL (Myocardial Ischemia Reduction with Acute Cholesterol Lowering), CARE (Cholesterol and recurrent Events), LIPID (Long-term Intervention with Pravastatin in Ischaemic Disease), 4S (Scandinavian Simvastatin Survival Study), HPS).
In the first outcome study comparing two statins, the PROVE-IT (Pravastatin or Atorvastatin Evaluation and Infection Therapy) study showed that high-dose atorvastatin (80 mg) was more effective than pravastatin (40 mg) at preventing further major cardiovascular events during the first 2-3 years after presentation with an acute coronary syndrome.
Typically, recommended evidence-based doses are simvastatin 40 mg nocte, pravastatin 40 mg od, and atorvastatin 10-80 mg od. Statins should not be prescribed to individuals with porphyria or severe liver or muscle disease, and caution is advised with their use in milder liver dysfunction, renal impairment, and in combination with fibrates.
Fibrates improve the lipid profile by activating peroxisome proliferatoractivated receptor alpha (PPAR-α), resulting in a mild lowering of TC and LDL-C.
Subjects with combined hyperlipidaemia, or other reasons for a low HDL and/or high triglycerides, respond well to fibrates.
The VA-HIT (Department of Veterans Affairs High-density Lipoprotein Cholesterol Intervention Trial) study showed that gemfibrozil (600 mmg bd) reduced major cardiovascular events in subjects with average cholesterol and low HDL (<1.0 mmol/L) levels.
Subgroup analysis of the HPS suggested that similar benefits were observed with simvastatin (40 mg) in such individuals.
For the majority of patients, fibrates are typically recommended as second-line agents in patients who are intolerant of statins, particularly if they have low HDL or high triglycerides.
The likelihood of liver or muscle side-effects is increased if a fibrate and statin are used in combination, and careful monitoring is recommended.
The risk is lower if the statin is combined with fenofibrate than with other fibrates.
reduced cholesterol and saturated fats (especially trans-fats)
increased plant stanols, sterols, and soluble fibre
adoption of Mediterranean diet.
cerebrovascular disease
renovascular disease
peripheral arterial disease.
 |
low HDL
high triglycerides (VLDL and remnant particles)
increased small, dense LDL
moderate hypertension
low-grade inflammation
a procoagulant state (increased PAI-1, and platelet activation)
increased oxidative stress
increased levels of harmful advanced glycation end-products.
Outcome is better when patients with diabetes and multivessel coronary disease undergo coronary artery bypass graft (CABG), rather than percutaneous revascularization (RITA (Radiofrequency Interstitial Tumour Ablation), BARI (Bypass Angioplasty Revascularization Investigation)).
Data from large studies suggest that the outcome after percutaneous coronary intervention (PCI) in patients with diabetes is improved by use of drug-eluting stents as well as abciximab, a platelet glycoprotein (GP)IIb/IIIa antagonist.
Visceral neuropathy is responsible for silent myocardial ischaemia, and reduced heart rate variability.
Diabetic nephropathy further contributes to cardiac risk by increasing BP and causing deterioration in the lipid profile.
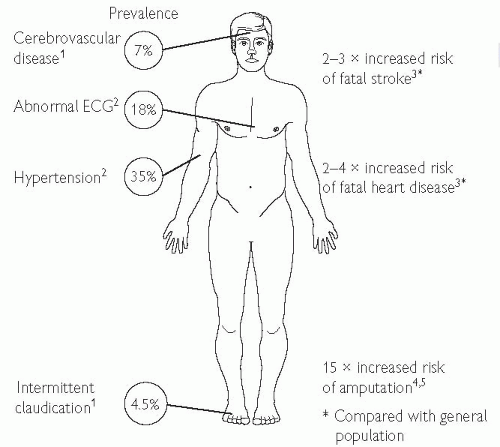 Fig. 5.7 Macrovascular disease in type 2 DM. 1 Wingard DL, Barrett-Connor EL, Scheidt-Nave C, McPhillips JB (1993). Prevalence of cardiovascular and renal complications in older adults with normal or impaired glucose tolerance or NIDDM. A population-based study. Diabetes Care 16: 1022-5. 2 UKPDS 6 (1990). Complications in newly diagnosed type 2 diabetic patients and their association with different clinical and biochemical risk factors. Diabetes Res 13: 1-11. 3 Balkau B, Pyörälä M, Shipley M et al (1997). Non-cardiovascular disease mortality and diabetes mellitus. Lancet 350: 1680. 4 King’s Fund (1996). Counting the cost. BDA. 5 Most RS, Sinnock P (1983). The epidemiology of lower extremity amputations in diabetic individuals. Diabetes Care 6: 87-91. |
Target glycosylated haemoglobin (HbA1c) is ≥7%.
Intense blood sugar control is better at reducing progression of microvascular disease and neuropathy than prevention of macrovascular events (UKPDS study).
Table 5.5 Therapeutic recommendations for CV prevention in diabetes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
It is characterized by:
visceral obesity
insulin resistance
moderate hypertension
dyslipidaemia (low HDL and high triglycerides with average cholesterol levels)
low-grade inflammation
Several definitions of this syndrome have been proposed, but of these the American National Cholesterol Education Program—Adult Treatment Panel (NCEP ATP-III) diagnostic criteria (see Table 5.6) are the most practical and widely used, as well as being a robust predictor of vascular risk.
The prevalence of this syndrome is increasing in parallel with the increase in numbers of obese and overweight individuals
It has been described in a high proportion of American children and adolescents.
It is projected to contribute substantially to the burden of atherosclerotic disease in society in the next few decades.
Table 5.6 ATP-III criteria for diagnosis of the metabolic syndrome | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
increases in blood glucose
adverse changes in blood lipids
increased levels of inflammatory cytokines such as tumour necrosis factor alpha (TNF-α), causing endothelial dysfunction
increases in vascular tone and BP
local proatherosclerotic changes in the vascular wall.
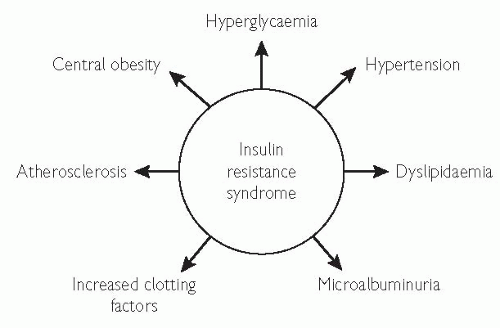 Fig. 5.8 Insulin resistance is an independent predictor of cardiovascular disease.1-4. 1 Haffner SM, Valdez RA, Hazuda HP, Mitchell BD, Morales PA, Stern MP (1992). Prospective analysis of the insulin-resistance syndrome (syndrome X). Diabetes 41: 715-22. 2 Haffner SM, Miettinen H (1997). Insulin resistance implications for type II diabetes mellitus and coronary heart disease Am J Med 103: 152-62. 3 Reaven GM (1994). Syndrome X: 6 years later. J Int Med 236(suppl 736): 13-22. 4 Abuaisha B, Kumar S, Malik R, Boulton AJ. (1998). Relationship of elevated urinary albumin excretion to components of the metabolic syndrome in non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 39: 93-9. |
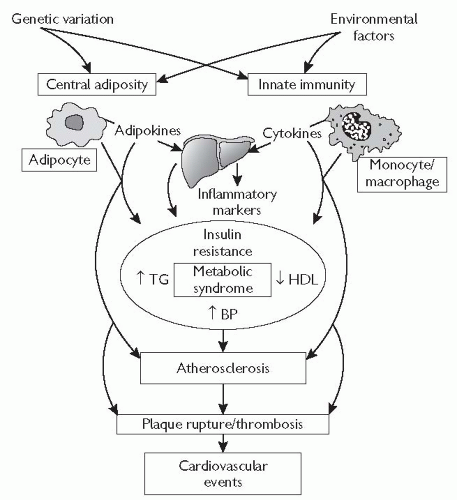 Fig. 5.9 Pathophysiology of atherosclerotic cardiovascular disease in the metabolic syndrome. Central adiposity and innate immunity play key roles in the development of insulin resistance, chronic inflammation, and metabolic syndrome features through the effects of adipokines (e.g. leptin, adiponectin, resistin) and cytokines (e.g. TNF-α, interleukin-6) on liver, skeletal muscle, and immune cells. In addition, monocyte/macrophage-, and adipocyte-derived factors may have direct atherothrombotic effects that promote the development of atherosclerotic cardiovascular events. Common genetic variants and environmental factors may impact the development of atherosclerosis at multiple levels, through influences on central adiposity, innate immunity, glucose and lipoprotein metabolism, and vascular function. TG = triglycerides. Reproduced with permission from Reilly MP, Rader J (2003). The metabolic syndrome: more than the sum of its parts. Circulation 108: 1546-51. |
The primary mode of action is to improve insulin sensitivity in muscle and adipose tissue (see Fig. 5.10).1,2
They are licensed as monotherapy, particularly in overweight patients with type 2 DM for whom metformin is not suitable.
They are associated with modest weight gain.3
They maintain lasting glycaemic control and, by targeting insulin resistance, improve a range of CV risk factors.4
There may be a role for this class of drugs in the prevention of DM: the TRIPOD study enrolled 266 Hispanic women, in S California at high risk of developing DM, and randomized them to the thiazolidenedione, troglitazone, or placebo for 30 months. There was a 50% reduction in risk of developing DM with troglitazone use, and the benefits were sustained for >6 months after discontinuing the drug.5
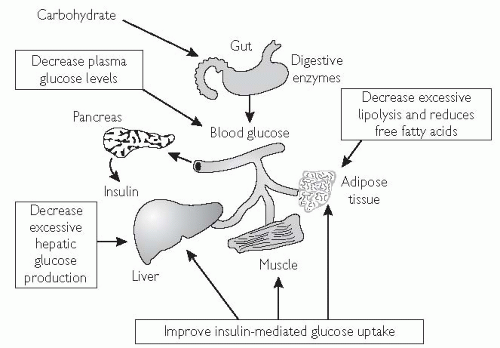 Fig. 5.10 Glitazones decrease insulin resistance at target tissues. |
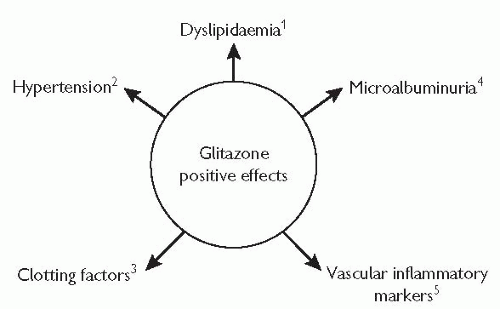 Fig. 5.11 Glitazones have positive effects on a range of cardiovascular risk factors. 1 Brunzell J et al (2001). Diabetes 50(suppl 2): Abs 567-P. 2 Bakris GL, Dole JF, Porter LE, et al (2001). Rosiglitazone improves blood pressure in patients with type 2 diabetes mellitus. Diabetes 49(suppl 1): A96, Abs 388. 3 Freed M et al (2000). Diabetologia 43(suppl 1): Abs 1024. 4 Bakris GL, Weston WM, Rappaport EB, et al (1999). Rosiglitazone produces long-term reductions in urinary albumin excretion in type 2 diabetes (abstract). Diabetologia 42(suppl 1): Abs 865 and poster. 5 Greenberg A, Haffner S, Weston W, et al (2001). Rosiglitazone reduces c-reactive protein, a marker of systematic inflammation, in type 2 diabetic patients. Diabetologia 44(suppl 1): A222, Abs 853. |
Angina pectoris is characterized by a deep and diffusely distributed central chest discomfort.
Certain features of pain are of discriminative value. Patients will not be able to point to where the pain is coming from with one finger, but will use an open palm or fist over the centre or left parasternal aspect of their chest.
The pain is not sharp, (some patients confuse ‘sharp’ with ‘severe’).
Pain lasts longer than a few seconds and rarely exceeds an hour without varying in severity. Most episodes will last 1-5 minutes.
The response to glyceryl trinitrate (GTN), if there is one, will be almost immediate.
Responses that take more than 5 minutes are unlikely to be related to the drug.
Chest wall tenderness suggests musculoskeletal pain and does not accompany angina.
Dyspnoea, fatigue, nausea, and recurrent belching may also represent underlying ischaemia and can occur in the absence of the classical central chest pain. The clue to underlying ischaemic heart disease (IHD) lies in their precipitation by exertion or emotional stress.
Stable angina—characterized by pain occurring after a relatively constant level of exertion.
Unstable angina—characterized by pain on minor exertion or at rest, which is either new onset or a dramatic worsening of existing angina.
Crescendo angina—characterized by pain on ever-diminishing levels of exertion, usually over a period of days.
Decubitus angina—provoked by lying flat.
Minimal limitation of ordinary activity. Angina occurs with strenuous, rapid, or prolonged exertion at work or recreation.
Slight limitation of ordinary activity; angina occurs on walking or climbing stairs rapidly; walking in cold, in wind, or under emotional stress.
Marked limitation of ordinary physical activity; angina occurs on walking 50-100 m on level ground or climbing 1 flight of stairs at a normal pace in normal conditions.
Inability to perform any physical activity without discomfort; angina symptoms may be present at rest.
This rarely reveals any direct evidence of IHD but will help with risk stratification and to exclude co-existing disease.
Measure the pulse rate. This may be slowed by inferior ischaemia due to atrioventricular (AV) node ischaemia. A resting tachycardia, if present, usually represents activation of the sympathetic nervous system but may be due to an arrhythmia precipitated by ischaemia.
BP measurement is essential to look for evidence of hypertension (predisposing to atheroma), or hypotension (may reflect cardiac dysfunction or over-medication).
Precordial examination should include palpation for LVH, cardiac enlargement, or dyskinesis, and auscultation for added heart sounds (heart failure or acute ischaemia), aortic stenosis or mitral regurgitation (due to papillary muscle dysfunction).
Examine for signs of heart failure by listening for fine, late-inspiratory crepitations at the lung bases, and looking for dependent pitting oedema (typically bilateral ankle ± leg oedema, but sacral oedema may be the only manifestation if the patient has been recumbent for some time).
Look for evidence of peripheral vascular disease by palpating for aortic aneurysm, feeling the carotid and limb pulses, listening for carotid, renal or femoral artery bruits, and assessing tissue integrity and capillary refill of the legs and feet.
Examine for signs of hypercholesterolaemia: the eyes for xanthelasmata and corneal arcus, and the skin and tendons (especially the Achilles) for xanthomata.
anxiety and hyperventilation
musculoskeletal chest wall pain
cervical or thoracic root pain
pneumothorax, pneumonia, or pulmonary embolus
oesophageal problem (inflammation/spasm)
other upper GI problem (gastritis, peptic ulcer, pancreatitis, cholecystitis)
pericarditis
aortic dissection
mitral valve prolapse
coronary emboli (LV mural thrombus, atrial myxoma).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


