Chapter 4 Connective Tissues of the Subendothelium
Varieties of Blood Vessels and Their Connective Tissue
The evolutionary transition from an open to a closed circulatory system is clearly reflected in the architecture of the blood vessels.1,2 The size and anatomical organization of individual vessels vary with their specific locations and functions in the body. The major vessels that carry blood directly from the heart are capable of storing and releasing large amounts of energy during the cardiac cycle. As a result, the walls of large arteries are relatively thick and more elastic to allow their expansion and contraction in response to the systolic and diastolic cycles of the heart. Without such elasticity, the intense surge in pressure as blood is ejected from the heart would inhibit its emptying, and the pressure in the vessels would fall too low for the heart to refill. The elasticity of large arteries enables them to store a portion of the stroke volume with each systole and discharge that volume with diastole. Thus, the unique structure of large arteries allows the flow of blood from the heart to be continuous, smooth, and efficient.
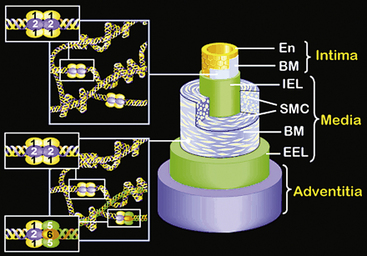
The walls of the large arteries contain three identifiable layers. The luminal surface of arteries contains a single layer of polygonal ECs connected by gap junctions. This cell layer rests on a basement membrane, which in turn is supported by a network of elastic fibers in a fenestrated plate called the internal elastic lamina. This region of the wall is called the tunica intima. The middle layer, called the tunica media, represents the bulk of the vessel wall, contains few elastic fibers but has a large number of VSMCs, with their long axes perpendicular to the lumen axis.3 Smooth muscle cells residing in the tunica media synthesize the major components of ECM that ultimately define the mechanical properties of the vessel. The extracellular space contains a variable mixture of collagen fibers in a continuous sheath adjacent to the elastic fibers. The external elastic lamina separates the medial and adventitial layers of the vessel wall. The outermost layer of the vessel wall, the tunica adventitia, consists primarily of collagen-rich ECM and the vasa vasorum, a network of vessels that supplies nutrients and O2 to the outer portion of arterial walls. Although the unique anatomy and high collagen content of the tunica adventitia help prevent arterial rupture at extremely high pressures, the adventitia is highly susceptible to vascular inflammation.
The walls of smaller arteries are intermediate in size. The tunica intima is relatively thin, as is the medial layer. The tunica adventitia of small arteries usually contains more densely packed collagen fibers arranged longitudinally along the vessel axis. Arterioles have simpler walls; their EC layer is surrounded by VSMCs, and the adventitia is smaller and more pliable compared with those of larger arteries.1,3 Capillaries adjoining the arterioles are surrounded by a few SMCs that control the amount of blood passing through them. The walls of arterial and venous capillaries are lined with flat ECs surrounded by a basement membrane; a discontinuous sheath of pericytes and a fibrous reticulum, made primarily of type III collagen, are attached to the basement membrane. The walls of venules also contain a reticular network of collagen fibers derived from type III collagen, along with smaller quantities of type I collagen fibers.
Vascular Morphogenesis and Extracellular Matrix
The preceding overview underscores the striking structural and phenotypic diversity of different branches of the vascular tree. Therefore it is not surprising that the vascular ECM displays similar complexity depending on its location in the vasculature.2–5 This caveat notwithstanding, all vascular ECM is composed of fibrillar and nonfibrillar components. The fibrillar component of the vascular connective tissue is mainly collagen, and a diversity of proteins and proteoglycans (PGs) make up the rest. What follows is an overview of the structural and functional properties of the major macromolecules that characterize the vascular ECM. For a more detailed discussion of the individual classes of ECM macromolecules, astute readers will need to consult specialized reviews and critical commentaries, a number of which are cited in the chapter.
Collagens
Twenty-eight genetically distinct types of collagen comprising 43 unique α chains have been identified in vertebrates (Table 4-1). The vast majority of these collagens exist in humans.6–9 Based on their domain organization and other structural features (Fig. 4-1), collagens may be categorized as (1) fibril-forming collagens represented by types I, II, III, V, XI, XXIV, and XXVII; (2) fibril-associated collagens with interrupted triple helices (FACIT; e.g., IX, XII, XIV, XVI, XIX, XX, XXI, XXII, and XXVI collagens); (3) collagens capable of forming hexagonal network (e.g., VIII, X); (4) basement membrane collagens represented by IV collagen; (5) collagens that assemble into beaded filaments (e.g., type VI); (6) anchoring fiber-forming collagens (e.g., VII); (7) plasma membrane-spanning types XIII, XVII, XXIII, and XXV collagens; and (8) collagens with unique domain organization, represented by types XV and XVIII.
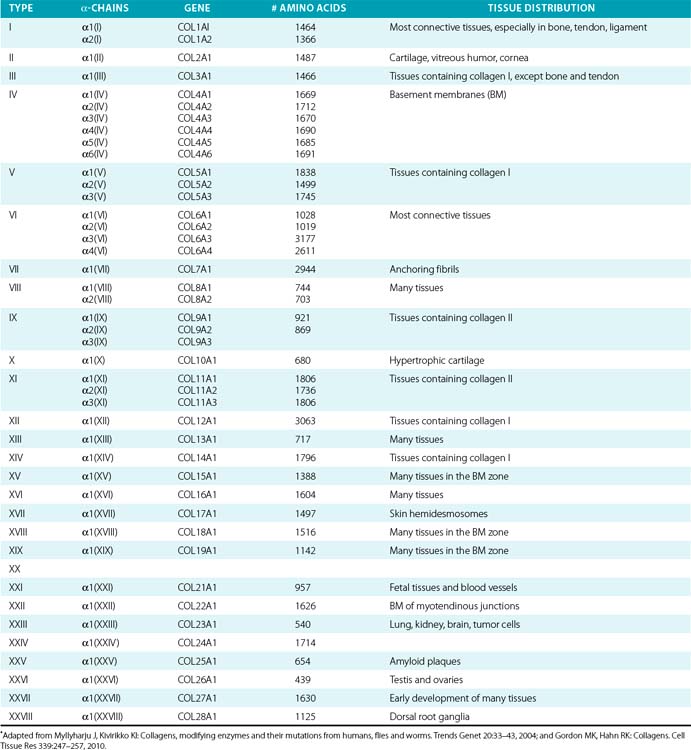
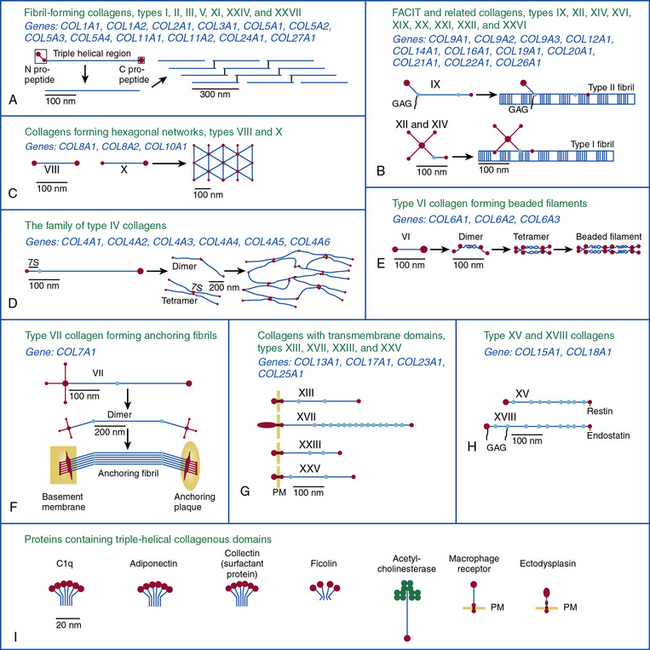
Figure 4-1 Classification of superfamily of vertebrate collagens.
(From Myllyharju J, Kivirikko KI: Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20:33–43, 2004.)
We should note that the nomenclature of proteins as collagens and their classification into different types is somewhat arbitrary, since collagen fibrils invariably consist of more than one type of collagen. For instance, type I collagen fibrils contain small amounts of type III, V, and XII; similarly, type II collagen fibrils contain significant amounts of collagen types IX and XI. Even more strikingly, types V and IX collagen are known to form hybrid fibrils. The discovery of collagens that have extensive non-triple-helical domains and several proteins that contain triple-helical domains, such as C1q, adiponectin, acetyl cholinesterase, and ectodysplasin (see Fig. 4-1), further challenge the notion of what constitutes a “true collagen” and how it should be classified. Although several collagen types are found in the vasculature, collagen types I and III are the dominant constituents of the blood vessel wall.6,7,9 Collagen types II and X are excluded from our discussion because they are not relevant to the ECM of the vascular endothelium.
Fibrillar Collagens
The collagen molecule, the basic unit of collagen fibers, has an asymmetrical, rodlike structure composed of three polypeptide chains called α chains. Because of the Gly-X-Y repeating units and their stereochemistry, each α chain forms a minor helix (Fig. 4-2). Three α chains wind around a common axis to form a right-handed triple helix. In some collagens, all three α chains are identical, while in others two or three unique α chains form the triple-helical molecule. Type I and type III collagens are the most abundant collagens in the blood vessel and together form the striated fibrils. With the exception of types XXV and XXVII, fibrillar collagens form an uninterrupted triple-helical domain of approximately 300 nm. The type I collagen α chains contain 338 Gly-X-Y repeats, and there are 341 such triplets in type III α chains. At both the NH2 and COOH ends of each α chain are short segments of nonhelical sequences of approximately 15 to 20 amino acid residues, referred to as telopeptides.
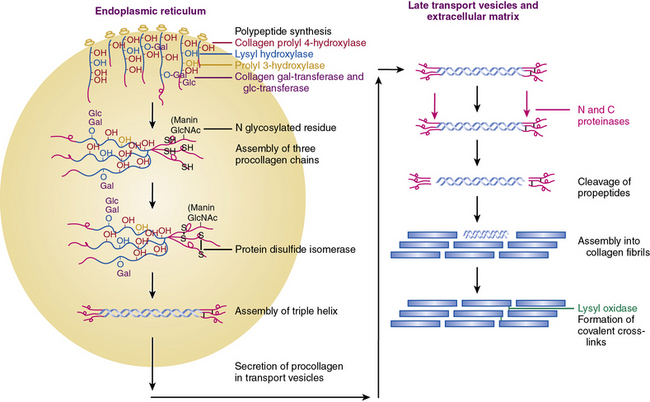
Figure 4-2 An overview of the main steps involved in the synthesis of fibril-forming collagens.
(From Myllyharju J, Kivirikko KI: Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20:33–43, 2004.)
The collagen triple helix is further stabilized by interchain hydrogen bonds contributed by hydroxyproline residues. Thus, the collagen molecule is a long cylindrical rod with dimensions of 1.5 nm × 300 nm. Under physiological conditions of ionic strength, pH, and temperature, collagen molecules spontaneously aggregate into striated fibrils. Fibril formation occurs by lateral aggregation of collagen molecules, in which each neighboring row of molecules is displaced along its long axis by a distance of 68 nm. In addition, within the same row, there is a gap of approximately 40 nm between the end of one molecule and the beginning of the next (see Figs. 4-1 and 4-2). The short nonhelical telopeptides at the NH2 and COOH ends of each α chain are located in the gap or hole zone of the fibril and are therefore accessible to enzymes that regulate collagen cross-linking.
Network-Forming Collagens
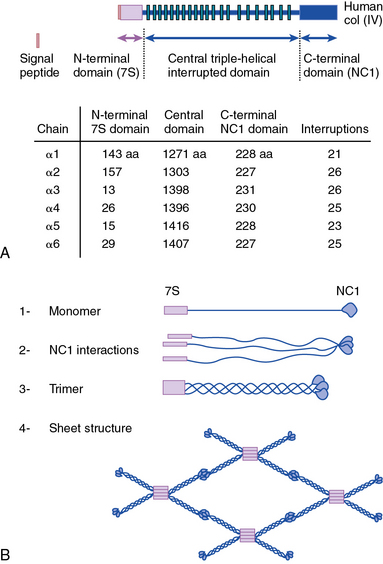
As shown in Figure 4-1, collagen types IV (α1-α6 chains), VI (α1-α5 chains), VIII (α1-α2 chains) and X are known to form networks in the ECM of basement membranes. The supramolecular organization and function of type IV collagen has been extensively characterized. Six different α polypeptide chains of collagen IV are each encoded by an evolutionary conserved gene. The amino and carboxyl propeptides of type IV collagen remain as integral parts of the molecules when they are deposited in the basement membrane. As a result, rather than forming a quarter-stagger, side-by-side alignment of individual molecules, as seen in types I, II, and III collagens, type IV collagen α chains form chicken-wire structures by end-to-end associations stabilized by lysine-derived cross-linking and interchain disulfide bonds (Fig. 4-3). The α1(IV) and α2(IV) collagen chains are more closely related to each other than to α3(IV)1, α4(IV), α5(IV), and α6(VI); the latter share a high degree of sequence homology with each other. The amino terminal domains of α1(IV) and α2(IV) collagen chains are 143 and 167 amino acids, respectively; the NH2-termini of the other four α chains are much smaller (ranging in size from 13 to 19 amino acids). Theoretically, all six α chains of type IV collagen may combine randomly to generate 56 unique triple-helical permutations. However, as shown in Figure 4-4, in vascular basement membranes the most common composition of triple-helical fibrils is [α1(IV)1]2 α2(IV). The [α3(IV)1]2 α4(IV) and [α5(IV)1]2 α6(IV) are also present in basement membrane.9,10
Type VI collagen, another network-forming molecule, is represented by six distinct α chains in the mouse and five α chains in humans; the gene encoding the putative α4(VI) collagen chain is not functional in humans. Heterotrimers of different α chains, encoded by unique genes, form the basic unit of type VI collagen. Alternate splicing of messenger ribonucleic acids (mRNAs) generates additional variants of α2 (VI) and α3 (VI) chains.7,9 The Gly-X-Y domains of α chains of type VI collagen microfibrils are rather short (about 330 amino acid residues) and are flanked by a number of von Willebrand factor (vWF) A domains.
Type VI collagen forms relatively unusual aggregates by a stepwise assembly into the triple-helical monomeric units that form dimers in an antiparallel fashion. The dimers in turn form tetramers, held together by disulfide bonds, to create scissors-like structures. The supramolecular assemblies of type VI collagen, formed by end-to-end associations of tetramers, appear as beads on a string, as revealed by electron microscopy.9 These characteristic structures have been observed in vascular subendothelium and skeletal muscle basement membranes. Type VI collagen microfibrils exhibit unique adhesive properties to other ECM components, such as other collagens, heparin, and vWF, and may be involved in the adhesion of platelets and SMCs. In the medial layer, type VI collagen facilitates interaction between SMCs and elastin by bridging the elastin fibers and cells.11
As illustrated in Figure 4-5 (see discussion in “Metalloproteinases”), types VIII and X collagens comprise a unique subfamily of collagens that form hexagonal networks. These relatively short collagens, containing noncollagenous domains on their NH2 and COOH termini, are collectively known as the multiplexin family of collagens. Type VIII collagen is expressed in many tissues, especially in the endothelium, while type X is exclusively associated with hypertrophic chondrocytes during cartilage and bone development. The preponderance of evidence to date indicates that the two α chains of collagen VIII, encoded by COL8A1 and COL8A2, assemble into homotrimers of α1(VIII) and α2(VIII) (Fig. 4-6). Hexagonal aggregates of type VIII collagen have been observed both in vivo (e. g., Descemet’s membrane of the cornea) and in vitro with purified protein. It is believed that type VIII collagen is capable of assuming other forms of macromolecular aggregates, since hexagonal lattices have yet to be observed in the subendothelial ECM.9
Fibril-Associated Collagens with Interrupted Triple Helices
As the name suggests, the FACIT collagens (types IX, XII, XIV, XVI, XIX, XX, XXI, XXII, and XXVI) do not form fibrils themselves but associate with other fibril-forming collagens.9 Type IX collagen, the prototype of this group, is cross-linked to the surface of type II collagen fibrils in cartilage (see Fig. 4-1); type XII and type XIV collagens are found in both cartilage and noncartilaginous tissues, where they are involved in controlling the diameter of collagen fibrils (see Fig. 4-1). The other FACIT-like collagens (e.g., types XVI, XIX, and XXII) are localized in specialized basement membranes. For instance, XVI collagen is associated with fibrillin 1 near the epidermal basement membrane. Collagen types XXI and XXII are closely related to each other in structure and are involved in formation of supramolecular aggregates in the basement membranes of myotendinous junctions.12,13 As a key constituent of cutaneous basement membranes, anchoring fibrils of type VII collagen form a structural continuum between the dermis and epidermis of normal human skin. The vWF A–like domain in collagen VII binds to fibrils of type I collagen in vitro.14
Minor Collagen Types with Unique Structures
As illustrated in Figure 4-1, the transmembrane collagens (types XIII, XVII, XXIII, and XXV) contain a cytoplasmic domain, a membrane-spanning hydrophobic domain, and extracellular triple-helical domains interspersed with noncollagenous domains; these collagens may also exist in a soluble form. Type XVII collagen is a unique member of this group that is expressed on the basal surface of keratinocytes that bind to laminin found in the basement membrane; compared with the other three members of this group, type XVII has a rather large intracellular domain whose function remains unknown. Collagen types XIII, XXIII, and XXV are similar to each other in their primary structure, but the patterns of their expression appear to be unique. Type XXV collagen is enriched in the senile plaques of Alzheimer’s disease brains.9,12,13,15 High expression of full-length collagen XXIII is found in the lungs, whereas its shed form is enriched in brain, suggesting that shedding of XXIII collagen occurs in a tissue-specific manner.
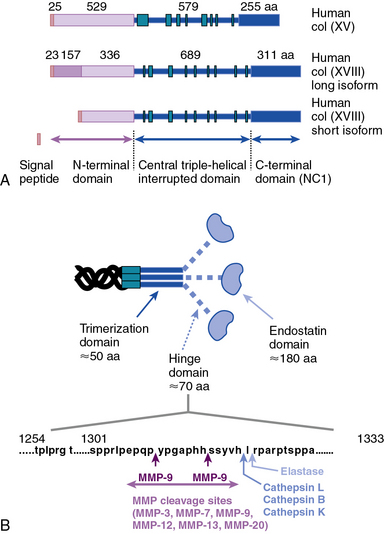
Collagen types XV and XVIII are highly pertinent to the EC biology in several ways.16,17 The full-length types XV and XVIII collagen are basement membrane components; their triple-helical domains share a high degree of homology. Collagen types XV and XVIII were initially identified as PG core proteins containing chondroitin sulfate and heparan sulfate (HS) side chains, respectively. The COOH-terminal domains of XV and XVIII collagens can be cleaved to generate biologically active peptides, endostatin and restin, respectively; these peptides inhibit migration of ECs and thus potently block angiogenesis. In vitro, recombinant collagen XV binds to fibronectin (FN), laminin, and vitronectin (VN) but not to fibrillar collagens, fibril-associated collagens, or decorin.18
Finally, collagens XXVI and XXVIII are newly discovered collagens that are unique both with regard to their structures and tissue-specific distributions. The triple-helical domain of type XXVI is rather small, with only 146 Gly-X-Y repeats. Expression of type XXVI collagen occurs predominantly in testis and ovary. The von Willebrand factor–A domains flank the triple-helical structure of type XXVIII collagen that is almost exclusively expressed in the peripheral nerves.19,20
Regulation of Collagen Biosynthesis
Collagen chains are synthesized as prepro-α chains from which the hydrophobic leader sequence is removed prior to secretion, and the pro-α chains are secreted into the extracellular space (see Fig. 4-2). The pro-α1(I) chain contains an NH2 propeptide (N-peptide) and a COOH propeptide (C-peptide). The N-peptide consists of a 139-residue sequence that precedes a 17-residue sequence of nonhelical telopeptide. This is followed by a 1014 amino acids-long Gly-X-Y helical sequence attached sequentially to a 26 residues-long COOH telopeptide and a 262-residues-long nonhelical C-peptide. The domain organization of pro-α2(I) and pro-α1(III) chains are similar except for minor variations in the number of amino acid residues.6,7,21
The genomic organization and chromosomal locations for genes that encode collagens have been studied. In humans, the genes encoding 43 distinct α chains are dispersed on at least 15 chromosomes. Unlike majority of the collagen-encoding genes, the six homologous α-chains of type IV collagen are encoded by genes that are located in pairs with head-to-head orientation on chromosomes 13 (COL4A1 and COL4A2), 2 (COL4A3 and COL4A4), and the X chromosome (COL4A5 and COL4A6). Interestingly, the promoters of these pairs of type IV collagens overlap, suggesting a coordinate regulation of the gene pairs. The precise molecular mechanisms of this regulation, however, remain incompletely known.6,7,9,21,22
The cis-acting elements of COLA1 and COLA2 genes are modularly organized on either side of the transcription start point (TSP). The regulatory elements are distributed over a distance of 100 to 150 kb of genomic DNA, depending on the specific gene and the assays used to study their transcriptional and posttranscriptional regulation. The tissue-specific and inducible activation of collagen genes involves complex interactions among the cis-acting modules of their promoters and enhancers. Promoters of COLA1 and COLA2 genes contain TATA boxes located 25 to 35 bp upstream of the TSP. Existence of a number of enhancer and repressor cis-elements around the TSP and in the first intron of COLA1 gene has been demonstrated. A key role for CAAT-binding factor, Sp1, Sp3, Ap1, nuclear factor (NF)-κB, and SMADs has been reported for several collagen genes; a number of orientation-dependent enhancer-like elements have also been documented.23,24
Fibrillar and nonfibrillar collagens found in subendothelial ECM are regulated by many cytokines and growth factors; collagen gene expression in response to cytokines (e.g., transforming growth factor [TGF]-β, tumor necrosis factor [TNF]-α, interleukins [ILs]), glucocorticoids, estrogen, androgen, and retinoids has been reported. The signaling cascades initiated by intrinsic and exogenous regulators impinge on a distinct set of cis-acting elements that bind to constitutive and inducible transcription factors. The emerging theme from these studies is that various cis– and trans-acting factors interact to recruit selective transcriptional coactivators and co-repressors in response to specific stimuli.23,24 However, the precise mechanisms that determine combinatorial interactions under physiological and inflammatory conditions remain to be elucidated.
Following translation, pre-procollagen α chains are chaperoned from the endoplasmic reticulum (ER) to the Golgi. It has been reported that the heat shock protein-47 (Hsp47) functions as a collagen-specific chaperone; thus, hsp47 is presumed to provide a quality control mechanism needed for proper maturation of newly synthesized procollagen chains. To demonstrate a role of hsp47 in vivo Nagai and coworkers25 inactivated Hsp47 gene by homologous recombination. The mutant embryos died in utero before 11.5 days of postcoitus development as a result of severely reduced levels of mature type I collagen in their tissues.
As shown in Figure 4-2, fibrillar and nonfibrillar collagens also undergo a number of posttranslational modifications for proper maturation; these include proteolysis of signal peptides, hydroxylation of key proline and lysine residues, glycosylation, and formation of interchain and intrachain disulfide bridges.6,7,21 Thus, optimal biosynthesis and assembly of collagens depends on a number of key enzymes. These include three hydroxylases, two collagen-specific glycosyl transferases, two unique proteinases that cleave the NH2– and COOH-termini, and a collagen-specific oxidase that is needed for cross-link formation. The posttranslational processing of the procollagen molecules also needs a peptidyl proline cis-trans isomerase and a protein disulfide isomerase (PDI).
Vitamin C–dependent 4-prolyl hydroxylase, an α2β2-tetramer located in the ER, plays a central role in collagen synthesis because 4-proline hydroxylation is obligatory for cross-link formation. In humans, there are three known isozymes of 4-prolyl hydroxylases, each with a distinct α subunit, but all contain PDI as their β subunit. Hydroxylation of lysine is carried out by lysyl hydroxylase, which also uses the same cofactors as prolyl hydroxylase and reacts only with a lysine residue in the Y position of the Gly-X-Y triplets. There are three known isozymes of lysyl hydroxylase in humans. The under-hydroxylation of procollagen leads to reduced secretion and rapid degradation. Deficiency of lysine hydroxylase is associated with skeletal deformities, tissue fragility, and vascular malformations.6,7,21
Assembly of procollagen chains into triple-helical molecules is directed by the COOH-terminal propeptide, with formation of interchain disulfide bonds (see Fig. 4-2). There is a high degree of structural conservation within the propeptide of fibrillar collagens across species. Following its triple-helical assembly, the procollagen molecule is secreted into the extracellular space. Once secreted, however, the NH2 and COOH propeptides are removed by the actions of N- and C-specific peptidases to yield the collagen molecule. The two proteinases that remove the NH2 and COOH propeptides from the newly synthesized collagen are represented by three isozymes each. The C-specific peptidases, members of the tolloid family, also cleave a number of other ECM proteins, and fragments of the propeptides can inhibit procollagen synthesis by a feedback mechanism.26,27
Extracellular Maturation of Collagens
During collagen fibril formation, lysyl oxidase catalyzes the oxidative deamination of specific lysine or hydroxylysine residues in the NH2– or COOH-terminal telopeptides to yield allysine and hydroxyallysine, respectively.28 These reactive aldehydes, being located in the hole zone of the fibril, are free to react with the ε-amino group of lysine or hydroxylysine residues on adjacent chains to form a Schiff base, which undergoes Amadori rearrangement to form ketoimine. With time, two ketoimine structures condense to form a trivalent cross-link, 4-hydroxy-pyridinium. All three types of cross-link may coexist in different fibrils.
A second type of cross-link seen in collagen originates from the condensation of two aldehydes in allysine or hydroxyallysine on adjacent chains. The resulting aldol condensate has a free aldehyde that reacts with other ε-amino groups of lysine or histidine, thus potentially linking three or four collagen chains.29 Once the aldehydes of allysine and hydroxyallysine are formed, subsequent aldamine and aldol condensation reactions proceed spontaneously. Thus, inter- and intramolecular cross-linking of fibrillar collagens results in formation of insoluble macromolecular aggregates that possess high tensile strength.
Metalloproteinases
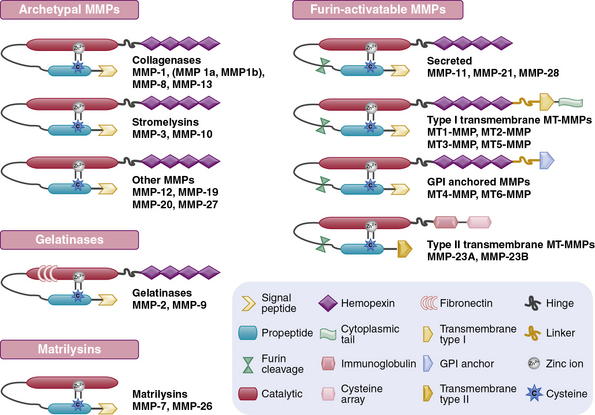
The structural and functional diversity of MMPs rivals that of the superfamily of collagens. The MMPs belong to a large family of zinc-dependent endopeptidases, the first of which was described nearly a half century ago. To date, the presence of 23 distinct MMPs has been reported in human tissues. Based on their cellular localization, these enzymes can be broadly subdivided into secreted and membrane-bound MMPs. However, a more detailed analysis of their structural organization and substrate specificities indicates that MMPs may be better classified as collagenases, gelatinases, stromelysins, metrilysins, and membrane-type MMPs.30–33
The architectural blueprint of a prototype MMP consists of three subdomains: the Pro-domain, the catalytic domain, and the hemopexin-like C-domain, connected to the catalytic domain via a short linker region (see Fig. 4-5). The catalytic domain of MMPs contains a Zn++ ion-binding amino acid sequence motif and a substrate-specific site. The prototypic MMP is synthesized as a pre-proenzyme and is maintained in latent conformation by the Pro-domain via interaction between a cysteine (located in the cysteine switch region of the Pro-domain) and Zn++ ion in the catalytic domain. Only when this interaction is disrupted, either by proteolysis of the Pro-domain or by a chemical modification of the cysteine, MMP becomes activated.32 A number of intracellular and extracellular proteinases, including other MMPs, are known to specifically degrade the Pro-domain to activate MMPs in vivo.
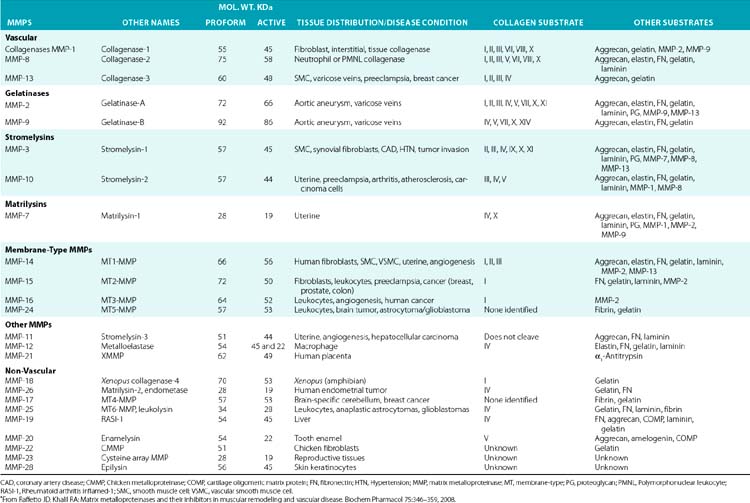
Although in vitro studies have identified numerous substrates for various MMPs (Table 4-2), the precise identities of their in vivo targets remain largely elusive. A number of macromolecules associated with ECM of the endothelium are potential in vivo targets of MMPs. For example, MMP-1 (collagenase 1) readily degrades collagen types I, II, and III, whereas MMP-8 (collagenase 2) digests types I, III, IV, V, VII, X, and XI collagen. Similarly MMP-2 (gelatinase A) degrades types I, III, IV, V, VII, X, and XI collagens, whereas gelatinase B (MMP-9) can degrade collagen types IV, V, XI, and XIV preferentially. MMP-13 (collagenase 3) is also capable of degrading collagens that are prevalent in subendothelial ECM (types I, III, VI, IX, and XIV). Many collagenous and noncollagenous ECM components are readily degraded by stromelysin-1 (MMP-3) and stromelysin-2 (MMP-10), whereas stromelysin-3 (MMP-11) does not degrade known collagens but readily breaks down laminin. Matrix metalloproteinases are also capable of digesting a number of other constituents of ECM, such as FN and elastin, and a variety of other cell- and ECM-associated molecules (see Table 4-2). The actions of some MMPs are likely to mediate highly regulated processing of ECM-bound pro-TGF-β and pro-IL-1.
Table 4-2 Members of the Matrix Metalloproteinase Family in Representative Vascular and Nonvascular Tissues*
Numerous studies have been undertaken to elucidate the molecular mechanisms by which the actions of MMPs are regulated in the tissues under physiological and pathological conditions.32 Two major mechanistic themes have emerged from these studies to explain the exquisite specificity of various MMPs. First, synthesis and localization of various pro-MMPs and their highly tissue-specific inhibitors (TIMPs) are regulated by autocrine and paracrine factors. Thus, cytokines such as IL-1 and TNF-α and a number of other circulating factors regulate expression of various MMPs at the transcriptional and posttranscriptional levels.
The second type of regulation of MMPs is exerted via the unique organization of their functional domains. As outlined earlier, the Pro-domain plays a critical role in maintaining the MMPs in a latent state that is altered by a number of physiological and pathological stimuli. Similarly, the presence of three cysteine-rich repeats, akin to those found in FN (see later discussion) in gelatinase A and gelatinase B, determines their affinities for elastin and collagen. The domain organization of MMPs allows them to be regulated by TIMPs; these inhibitors reversibly bind to MMPs in a 1:1 stoichiometry and inhibit enzymatic activity.34 Tissue inhibitors of MMPs, represented by four homologous proteins (TIMP1 to 4), preferentially inhibit various MMPs.35,36 For example, whereas TIMP3 potently inhibits MMP-9, both TIMP2 and TIMP3 inhibit membrane-type 1 (MT1)-MMP. In contrast, TIMP1 is a very poor inhibitor of MT-3-MMP but a potent inhibitor of MMP-3.34
Concerted actions of various MMPs and their TIMPs regulate key events in the formation of blood vessels in the developing embryo, and the processes of neovasculogenesis and angiogenesis in the adult in response to injury and regeneration (Table 4-3). Formation of new blood vessels from existing vessels is dependent on extensive turnover of subendothelial ECM. This process enables migration of blood vessel–associated cells, liberation of angiogenic factors sequestered in the ECM, and exposure of cryptic cell-regulatory domains found in the intact fibrillar and nonfibrillar components of connective tissue. Therefore, a crucial balance between MMPs and TIMPs is essential for maturation of newly formed blood vessels and ongoing maintenance of their structural integrity. These processes are known to play a critical role during embryogenesis; the formation of solid tumors and their acquisition of invasive, metastatic phenotype is also vitally dependent on the emergence of new blood vessels.37 MMP-2 binds to the αvβ3 integrin and promotes angiogenesis and tumor growth.38 In contrast, the transmembrane MMP, MT1-MMP, cleaves αvβ3 integrin and enhances its affinity for its ligands containing arginine-glycine-aspartic acid (RGD) sequences.
Table 4-3 The Effect of Matrix Turnover on Vascular Pathologies*
| Model | Effects | |
|---|---|---|
| Aneurysm | MMP-3−/− / ApoE−/− | ↓ Aneurysm |
| MMP-9−/− | ↓ Aneurysm | |
| MMP-12−/− | ↔ Aneurysm | |
| Broad-range MMP inhibitor LDLR−/− | ↓ Aneurysm | |
| TIMP-1−/− / ApoE−/− | ↑Aneurysm | |
| TIMP-1 ↑ rat | ↓ Aneurysm | |
| Neointima formation | MMP-9 ↑rat | ↑SMC migration ↓ matrix content ↑Luminal diameter |
| Broad-range MMP inhibitor | ↓ Early and ↔ late neointima formation | |
| LDLR−/− Doxycycline, MMP inhibition rat | ↓ Neointima formation | |
| TIMP-1 ↑human vein | ↓ Neointima formation | |
| TIMP-2 ↑human vein | ↓ Neointima formation | |
| TIMP-3 ↑human and pig veins | ↓ Neointima formation | |
| MMP-9−/−, mouse carotid ligation | ↓ Intimal hyperplasia,↑ collagen content | |
| Remodeling | MMP-12 ↑ | ↓ Luminal diameter |
| MMP inhibitor pig | ↓ Constrictive remodeling | |
| Atherosclerosis | MMP-1 ↑/ ApoE−/− | ↓ Plaque size ↓ collagen content |
| MMP-3−/− / ApoE−/− | ↑ Plaque size ↑ collagen content | |
| MMP-3 ↓ human, promoter polymorphism | ↑Plaque progression | |
| MMP-9 ↑ human, promoter polymorphism | ↑Triple-vessel disease | |
| MMP-9 ↑ human promoter polymorphism | ↔ Coronary artery stenosis | |
| Broad-range MMP inhibitor LDL−/− | ↔ Plaque size | |
| TIMP-1−/− / ApoE−/− | ↓ Plaque size ↑ lipid core content | |
| TIMP-1−/− / ApoE−/− | ↔ Plaque size, medial rupture, micro aneurysms | |
| TIMP-1 ↑/ ApoE−/− | ↓ Plaque size ↑ collagen content | |
| TGF-β inhibition ApoE−/− | ↑ Plaque vulnerability, intraplaque hemorrhage |
MMP, matrix metalloproteinase; Apo, apolipoprotein; LDLR, LDL receptor; TIMP, tissue inhibitor of matrix metalloproteinase; SMC, smooth muscle cell; TGF, transforming growth factor; +/+, transgenic overexpressing mice; −/−, knock-out or homozygous deficient mice; ↑, upregulation or increased; ↓, downregulated or decreased.
*Adapted from Heeneman S, Cleutjens JP, Faber BC, et al: The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J Pathol 2003:516, 2003.
Elastin
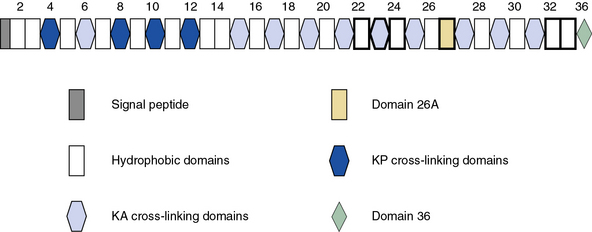
Blood vessels are endowed with a high degree of elasticity, and subendothelial elastic fibers are responsible for the resilience of the vasculature to cycles of deformity and passive recoil during diastole and systole, respectively. The elastic fiber consists of an insoluble core of polymerized tropoelastin surrounded by a mantle of microfibrils. A schematic representation of the modular organization of human tropoelastin is shown in Figure 4-7. The primary structure of tropoelastin consists of hydrophilic and hydrophobic domains; these may be further divided into subdomains based on the composition of their amino acid sequences (see Fig. 4-7). The mechanical properties of the elastic fiber are similar to rubber (i.e., the degree of elongation without irreversible changes per unit force applied to unit cross-sectional areas is high).
Organization of the elastic fibers has been studied by electron microscopic, biochemical, and genetic approaches, and a number of key insights have been gathered in recent years.3 Elastin is a major constituent of the elastic fiber and may contribute as much as 50% of the dry mass of large arteries.39 The elastic fibers begin to form at mid-gestation by deposition of tropoelastin, the soluble precursor of the cross-linked mature elastin, on a template of fibrillin-rich microfibers. The cross-linked elastin contained in the elastic fibers produced during late fetal and postnatal development generally lasts a lifetime.
Elastin has an amorphous appearance in the electron microscope; microfibrils appear as 10- to 15-nm diameter filaments. The assembly of elastic fibers occurs via a stepwise process that includes formation of a scaffold of microfibrils that facilitate deposition of tropoelastin monomers (Fig. 4-8), followed by extensive cross-linking to form the functional polymer.3,40,41 Tropoelastin is the soluble monomer of elastin that is one of the most apolar and insoluble proteins in nature. Although the glycine and proline content of elastin is similar to fibrillar collagens, elastin contains no hydroxyproline or hydroxylysine, and very small amounts of polar amino acids. Elucidation of the molecular organization of elastin has been difficult because of the technical problems in obtaining large quantities of tropoelastin. Therefore, scientists have relied mainly on the structure of fragments of hydrolyzed soluble elastin and recombinant tropoelastin produced in bacteria.
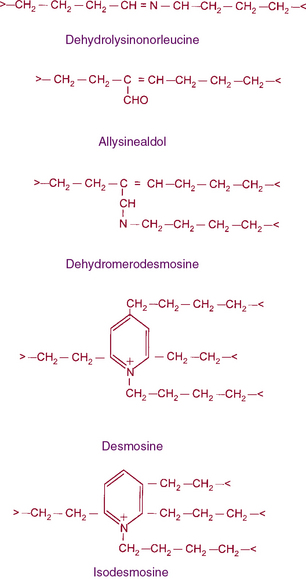
Figure 4-8 The structures of cross-links found in elastin.
Desmosine and isodesmosine represent final products of lysine-derived cross-links.
As illustrated in Figure 4-7, human tropoelastin is encoded as a 72-kD polypeptide that is characterized by a series of tandem repeats. The tropoelastin amino acid sequence is divided into hydrophobic domains that are rich in nonpolar amino acids (glycine, valine, and proline) that typically occur as repeating units; these sequences alternate with hydrophilic domains that are enriched in lysine and alanine. In vitro, elastin undergoes a process of ordered self-aggregation called coacervation (aligning and concentrating the protein in unit spheres) prior to cross-linking. Tropoelastin binds to cell surface glycosaminoglycans as well as to αvβ3 integrins.42 Although the sequential interactions of tropoelastin with fibrillins and its associated molecules are poorly defined, it is thought that the process of elastic fiber assembly is initiated on the cell surface.43,44 This is caused by specific interactions of the individual hydrophobic domains of tropoelastin, since it has an intrinsic ability to organize into polymeric structures.39
In vivo, tropoelastin probably interacts with microfibrils prior to aggregation and becomes cross-linked by lysyl oxidase.40,41,45 Soluble precursors of elastin are not found in extracts of normal tissues. This provides a clue as to the rapid formation of mature, highly cross-linked elastin fibers and the low rate of tropoelastin synthesis. In experimental conditions such as copper deficiency or lathyrism induced by β-aminopropionitrile, which inhibits lysyl oxidase activity and thus cross-link formation, a soluble 72 kD tropoelastin can be extracted from the aorta. Like collagens, newly synthesized tropoelastin undergoes posttranslational modifications before its assembly into elastic fibers; in fact, the same lysyl oxidase reacts with both collagens and elastin.46 In contrast to collagen, however, reduction of double bonds in the elastin cross-link occurs spontaneously, and the quantity of lysine involved in cross-linking is much larger in elastin than in collagen (see Fig. 4-8). Oxidative deamination of lysine residues, followed by subsequent condensation reactions, creates the unusual cross-links found in elastin. All of the cross-links in elastin are derived from lysyl residues through allysine (Fig. 4-9). However, the precise molecular reactions needed to form desmosine remain to be elucidated. Cross-linking in elastin occurs frequently, not only between peptide chains but also within the same polypeptide chain, producing intrapolypeptide links. The cross-linking process is highly efficient, and it is unclear how the cross-linking sites in the monomer get aligned.
Genomic organization of the tropoelastin gene indicates that functionally distinct cross-linking and hydrophobic domains of tropoelastin may be encoded by distinct exons. Short segments rich in alanine and lysine are clustered to apparently delimit the cross-linked region. These amino acids are clustered in the α-helical configuration of tropoelastin, where each begins with tyrosine followed by Ala-Ala-Lys or Ala-Ala-Ala-Lys. In humans, several distinct tropoelastin polypeptides may be generated by alternative splicing (see Fig. 4-7). Space-filling atomic models indicate that lysines separated by two or three alanyl residues in α-helical conformation protrude on the same side of the helix. Hence, the sequence Lys-Ala-Ala-Lys allows formation of dehydrolysinonorleucine, whereas the sequence Lys-Ala-Ala-Ala-Lys accommodates either aldol condensation or dehydrolysinonorleucine formation. Condensation of the two intrachain cross-links could result in the formation of the interchain desmosine cross-links. The alanine- and lysine-rich cross-linking segments are separated by large hydrophobic segments of 6 to 8 kD, which are in a β-spiral structure with elastomeric properties. Within the hydrophobic segments, a repeating pentapeptide (Pro-Gly-Val-Gly-Val) is present. A collagen-like sequence (Gly-Val-Pro-Gly) occurs quite frequently, which would explain the limited susceptibility of tropoelastin to bacterial collagenase (Pro-Gly-X-Y). The sequence Gly-X-Pro-Gly is recognized by the prolyl hydroxylase involved in the cross-linking of collagens (see earlier discussion).
Elastin Metabolism and Vascular Homeostasis
After deposition, tropoelastin production is strikingly reduced; the half-life of elastin in normal humans has been estimated in years. In the event of injury, production of elastin can be quickly initiated. A number of growth factors and cytokines induce biosynthesis of tropoelastin. Under these conditions, a very specific set of proteinases named elastases are responsible for elastin remodeling. Elastin fibers may be degraded by a number of MMPs, particularly MMP-2, -3, -9 and -12, that are present as latent enzymes under physiological conditions but are activated following vessel wall injury.47 The MMPs from neutrophils or macrophages are believed to degrade the elastin-rich ECM found in inflamed tissues. A hereditary defect in circulating elastase inhibitors is associated with a progressive destruction of the elastin-rich alveolar wall, resulting in premature emphysema. Furthermore, experimental instillation of elastase into the lungs of animals causes destruction of the lung similar to that seen in patients with α1-proteinase inhibitor deficiency.
Mice with a disrupted elastin gene have provided important insights into the function of elastin protein. Heterozygous (elastin+/− mice) had decreased arterial compliance and were hypertensive. The homozygous elastin-null mice died young due to arterial obstruction caused by uncontrolled proliferation of smooth muscle cells (SMCs).39,48 A direct link between occlusive vascular diseases and perturbation in the organization of the elastic fibers in the vessels has also been established.49 Mutations in the elastin gene are associated with supravalvular aortic stenosis (SVAS) and Williams-Beuren’s syndrome (WBS), pediatric disorders characterized by hemodynamic stress and loss of elasticity.49 Furthermore, haploinsufficiency of elastin resulting from aberrant degradation of mutated protein in humans or ablation of the elastin gene in transgenic mice caused intimal hyperplasia and thickened arteries.40,45,50–52 Apparently, VSMCs, the primary producers of elastin, organized more cell layers to compensate for lost elasticity and biomechanical support in developing blood vessels of elastin haploinsufficient patients and transgenic mice.
Vascular smooth muscle cells from SVAS patients, WBS patients, and elastin−/− mice show increased rates of proliferation and chemotactic migration, and reduced rates of elastin synthesis in vitro.50–52 Exogenous supplementation of recombinant tropoelastin and α-elastin to these cultures reversed their phenotype. The elastin-rich ECM serves as an autocrine regulator of VSMC; Karnik et al.53 inserted elastin-coated stents in a porcine coronary injury model of restenosis and found that intimal thickness and arterial stenosis were significantly reduced. Although the identities of the specific receptors mediating elastin VSMC interactions and the signaling mechanisms underlying vascular remodeling remain obscure,54 restoring elastin to an injured arterial wall is known to reduce obstructive vascular pathology.55,56 Inhibitors of MMPs have been shown to prevent degradation of elastic fibers after vascular injury and ameliorate neointimal thickening.47
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


