Congenital Heart Disease in Adults: Introduction
Congenital cardiac anomalies are the most common birth defects in humans, affecting approximately 0.8 in 100 live births. While the incidence of congenital heart disease is expected to decline as a consequence of improved prenatal diagnosis, the number of patients surviving with congenital heart disease, both in the United States and worldwide, has increased significantly over the past three decades. Over 85% of infants born with cardiovascular anomalies now can expect to reach adulthood. Reduced mortality rates can be attributed to improved diagnostic abilities, enhanced surgical and nonsurgical therapies, and improvements in intensive care. For the first time, the number of adults with congenital heart disease exceeds the number of children with the disorders.
The increase in the number of adults with congenital heart disease requires that the physicians managing the care of these patients have improved knowledge of simple and complex anatomy and physiology. Although actual numbers are difficult to ascertain, it has been estimated that approximately 1 million adults in the United States alone currently have congenital heart disease and that the number of patients reaching adulthood with treated congenital heart disease will increase by approximately 9000 per year.
These patients fall into several broad categories: those surviving into adulthood without intervention and perhaps without clinical recognition, those surviving with curative surgical or nonsurgical intervention, and those surviving with palliative surgical or nonsurgical intervention. Nonsurgical interventions may include catheter-based valvuloplasty, stenting, coiling, or device occlusion. The patients who are today making the transition into the adult congenital heart disease population have hemodynamic and cardiac problems differing from those in previous eras. Surgical techniques have evolved, intervention occurs earlier and is often definitive rather than palliative, and a greater number of patients with complex single-ventricle physiology and various modifications of cavopulmonary anastomoses (Glenn shunt, Fontan procedure) will reach adulthood. Because of the unique clinical conditions and needs of these patients, the American College of Cardiology and the American Heart Association have recommended that all patients with moderate or complex congenital heart disease be evaluated at least once by a physician with specialized training and expertise in adult congenital heart disease. Furthermore, it is recommended that diagnostic and interventional cardiac catheterization and electrophysiologic and surgical procedures in these patients be performed in regional adult congenital heart disease centers.
Acyanotic Congenital Heart Disease
The most common acyanotic congenital heart defects include abnormalities of the heart valves and great vessels, ventricular or atrial communications with left-to-right shunting, and such lesions as partial anomalous pulmonary veins and anomalous coronary arteries.
- History of murmur since infancy, coarctation repair, or endocarditis.
- Early systolic ejection click, harsh crescendo-decrescendo systolic, or early decrescendo diastolic murmur.
- Left ventricular hypertrophy.
- Abnormal bicuspid or dysplastic aortic valve with stenosis or regurgitation on Doppler echocardiography.
Congenital aortic stenosis is the most common anomaly encountered in the adult population and constitutes approximately 7% of all forms of congenital heart disease. The male:female ratio is approximately 2–3:1. The term “bicuspid aortic valve” is actually a misnomer; a raphe caused by commissural fusion of two leaflets usually exists. The valve is often dysplastic, with thickened, rolled, and calcified leaflets. The predominant pathophysiology results from mildly obstructed nonlaminar (disturbed) flow across the abnormal valve. A left ventricle-to-aorta pressure gradient of variable severity occurs, setting the stage for the inevitable deterioration of the valve with long-term calcium deposition and progressive stenosis or regurgitation. The valve is also at risk for endocarditis, which can lead to early destruction and regurgitation. Congenital aortic valve disease frequently occurs as a developmental “triad” with coincident aortopathy and coarctation, and these conditions should be sought clinically and echocardiographically in patients with congenital aortic valve disease. Familial congenital aortic valve disease occurs occasionally. While the genetics of this are incompletely understood, mutations in the gene encoding Notch 1 have been implicated in a subset of these patients.
The individual with congenital aortic stenosis is usually asymptomatic unless hemodynamically significant stenosis or regurgitation is present. Routine physical examination reveals a normal carotid pulse contour and left ventricular (LV) impulse, a normal S2, an early systolic click or sound, and an early-peaking systolic murmur. Identifying these patients in the asymptomatic stage is important. Current guidelines do not recommend endocarditis prophylaxis for the native valve. Refraining from high-level isometric exercise is generally believed to preserve valve function and limit valve regurgitation. The presence of a diastolic murmur of aortic regurgitation in a patient with a febrile illness should alert the clinician to the possibility of endocarditis and prompt the performance of appropriate diagnostic tests (eg, blood cultures, echocardiogram).
Congenital aortic stenosis is progressive; once hemodynamically significant valvular disease develops in a patient, generally in the fifth or sixth decade of life, the symptoms and signs are identical to those of a patient with acquired aortic valvular disease. Dyspnea, chest pain, and exertional syncope are the classic presenting symptoms. When stenosis predominates, the carotid upstroke is delayed and diminished in volume, the systolic click is no longer present, S2 is single, and the systolic murmur is crescendo-decrescendo, peaking in late systole. The murmur of aortic regurgitation is often present. The finding of upper extremity hypertension should alert the examiner to the possibility of concomitant aortic coarctation.
The major electrocardiographic (ECG) findings occur in the presence of hemodynamically significant disease and include left ventricular hypertrophy (LVH) with high QRS voltage, left-axis deviation, and repolarization changes; left atrial enlargement may also be present. The chest radiographic findings are nonspecific. With predominant valvular aortic stenosis, the cardiothoracic ratio may be normal, but LV enlargement and calcification may be evident in the region of the aortic valve on the lateral film. A dilated ascending aorta or a prominent aortic knob may be seen. The cardiac silhouette is enlarged in patients with predominant aortic regurgitation. Pulmonary vasculature may be prominent in the presence of congestive heart failure (CHF).
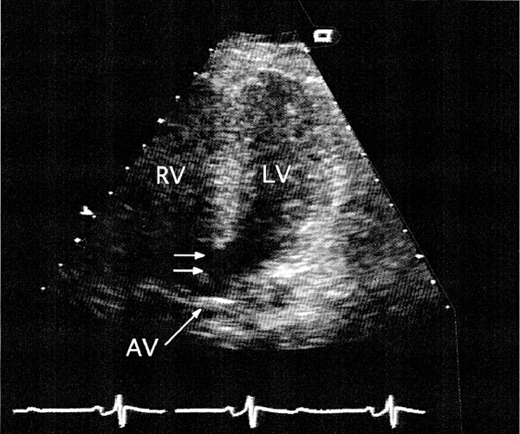
In the child and younger adult, the abnormally thickened leaflets of the congenitally abnormal valve are readily seen, and the bicuspid valve with its ovoid appearance in systole is apparent (Figure 31–1). On M-mode echocardiography, the point of closure may be eccentric. Heavy calcification often obscures the original valve morphology in the older individual with stenosis. The peak systolic gradient in severe aortic stenosis is usually greater than 64 mm Hg (peak velocity > 4 m/s by continuous wave Doppler). Aortic valve area can be accurately calculated by the continuity equation. Severe aortic stenosis is defined as a valve area of less than 1.0 cm2 or 0.5 cm2/m2. The LV shows concentric hypertrophy with thick walls and normal cavity dimensions, and the LV ejection fraction is usually normal. In cases with reduced ejection fraction, however, the peak gradient across the aortic valve is generally lower.
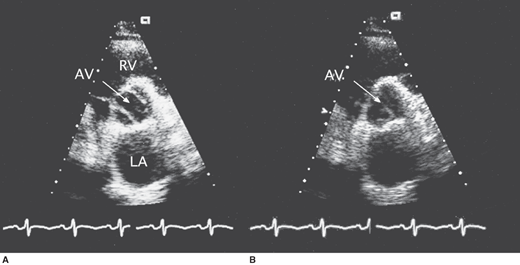
In hemodynamically significant aortic regurgitation, the high-velocity diastolic color-flow jet is broad at its site of origin below the aortic valve. Spectral Doppler imaging demonstrates a dense diastolic velocity signal with a short pressure half-time (< 400 ms), and diastolic flow reversal can be recorded in the descending aorta. The LV shows eccentric hypertrophy with normal LV wall thickness and a dilated cavity.
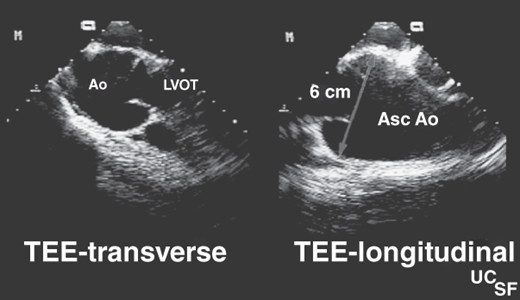
In occasional patients with poor precordial windows, transesophageal echocardiography (TEE) may be required to define valve anatomy by demonstrating commissural fusion and asymmetric sinuses of Valsalva. Systolic doming of the leaflets can be easily seen in the long axis view of the LV outflow tract, which also allows measurements to be taken of the aortic valve annulus, sinuses of Valsalva, sinotubular ridge, and ascending aorta.
Cardiac magnetic resonance angiography (MRA) is a possible alternative imaging technique.
Indications for cardiac catheterization in congenital aortic valve disease have changed significantly because most of the diagnostic data are now available noninvasively. According to current guidelines, however, cardiac catheterization is indicated when noninvasive tests are inconclusive or when there is a discrepancy between noninvasive tests and clinical findings regarding severity of aortic stenosis. It is also indicated preoperatively in patients at risk for atherosclerotic coronary artery disease (men age 35 years or older, premenopausal women age 35 years or older who have coronary risk factors, and postmenopausal women) and in patients for whom a pulmonary autograft (Ross procedure) is contemplated and if the origin of the coronary arteries was not identified by noninvasive techniques.
Serial imaging with magnetic resonance imaging (MRI) permits accurate and reproducible follow-up of aortic dilatation and is accepted as the standard of care for follow-up of repaired coarctation, a frequent association with a bicuspid aortic valve.
Valvular aortic stenosis should be distinguished from subaortic stenosis, which may be due to an obstructing fibrous membrane in discrete subaortic stenosis, which is more frequently encountered in adults, or by a tubular fibromuscular channel that usually presents in childhood. Aortic regurgitation, commonly associated with discrete membranous subaortic stenosis (in approximately 60% of cases), increases in frequency with age. Regurgitation may occur due to leaflet thickening induced by direct trauma to the aortic leaflets from the high-velocity jet or by interference with leaflet closure from the membrane. The aortic valve appearance and the severity and mechanism of aortic regurgitation must be carefully assessed.
Recurrent stenosis caused by regrowth following surgical resection of the fibromuscular ridge is sometimes encountered in the adolescent or adult. Discrete subaortic stenosis and congenital valvular aortic stenosis must also be distinguished from dynamic LV outflow-tract obstruction caused by hypertrophic cardiomyopathy (see Chapter 23). Supravalvular aortic stenosis is frequently seen in patients with Williams syndrome.
When aortic regurgitation is the predominant lesion and ascending aorta dilatation is present, the condition must be distinguished from Marfan syndrome and other genetic aortopathies. This latter condition is characterized by dilatation of the aortic root at the level of the sinuses of Valsalva (Figure 31–2). The aortic valve leaflets are not thickened, and regurgitation is caused by failure of leaflet coaptation caused by the root dilatation. With valvular stenosis, the aorta narrows toward normal at the sinotubular junction, and the descending thoracic aorta is spared. In some patients with a bicuspid aortic valve, an underlying abnormality of the medial layer of the aorta above the valve predisposes to the dilatation of the aortic root, which may progress to aneurysm formation or rupture and places the patient at risk for aortic dissection. All components of the vessel wall, smooth muscle, elastic fibers, collagen, and ground substance can be affected and should be recognized as a potential risk in the surgical patient.
The natural history of aortic stenosis presenting in childhood depends largely on the severity of the stenosis at the time of diagnosis. During a 25-year follow-up period, approximately one-third of the children with a peak systolic gradient of less than 50 mm Hg who were treated medically, required surgery, in contrast to 80% of those with an intermediate gradient (50–79 mm Hg). Of those treated surgically for a gradient of more than 79 mm Hg, approximately one-fourth required reoperation; reoperation was more common in those treated with initial valvotomy (30%) than aortic valve replacement (5%). The overall 25-year survival rate is approximately 85%; sudden death accounts for approximately half of the cardiac-related deaths.
Once symptoms of aortic stenosis develop, the prognosis without valve replacement is poor; the 5-year mortality rate is approximately 90%. Although percutaneous valvuloplasty has been successful in children and adolescents, the results in adults (even those with congenitally abnormal valves) have been disappointing. Therefore, surgery with aortic valve replacement, rather than percutaneous valvuloplasty, is generally mandated. Surgery is indicated in the symptomatic patient with a valve area of less than 1.0 cm2 (or < 0.5 cm2/m2). It should be considered in the asymptomatic patient with critical stenosis when the patient requires cardiac surgery (eg, coronary artery bypass surgery) for another lesion. A particularly difficult management decision ensues in the asymptomatic woman with severe aortic stenosis who is contemplating pregnancy. Valve replacement prior to pregnancy should be considered when there is evidence of LV dysfunction or reduced exercise tolerance on objective stress testing.
Patients with a bicuspid aortic valve and concomitant annuloaortic ectasia may show a more rapid progression of aortic regurgitation and require surgical intervention earlier than those patients with pure aortic stenosis.
An ideal substitute for replacing the aortic valve does not exist. Homografts and bioprosthetic valves can develop rapid calcific degeneration, causing valve dysfunction, particularly in the younger cohort of patients. Mechanical valves, although extremely durable, require anticoagulation to reduce the complication of thromboembolism. The risks associated with long-term anticoagulation have made surgical options to avoid the use of mechanical valves desirable alternatives. This is particularly germane to the choice of prosthetic valve in young women of childbearing age, in whom management of anticoagulation can be problematic. The Ross procedure (in which the autologous pulmonary valve replaces the aortic valve, and an aortic or pulmonary homograft replaces the pulmonary valve) has been increasingly performed for a variety of LV outflow tract diseases, including aortic insufficiency and valvar aortic stenosis with or without other forms of obstruction (eg, subaortic stenosis, supravalvar stenosis, and arch hypoplasia). Although the Ross procedure is more complex than simple aortic valve replacement, it can be performed with a low mortality rate in selected patients. Advantages of the pulmonary valve autograft include freedom from anticoagulation and the absence of compromise from host reactions and autograft growth, making it an attractive option for aortic valve replacement in infants and children. It is recognized, however, that the pulmonary homograft will require replacement for degenerative disease and size restriction in children. In adults who are confronting surgery for a stenotic aortic valve in the fifth or sixth decade of life, the Ross procedure has shown to be an acceptable alternative to the usual mechanical or bioprosthetic valve. However, recently recognized problems associated with the Ross procedure include progressive dilatation of the neo-aortic root, pulmonary conduit stenosis, and neo-aortic valve regurgitation. Contraindications to the Ross procedure include advanced three-vessel coronary artery disease, poor LV function, a severely calcified or dilated aortic root, or pulmonary valve pathology.
- History of systolic murmur since infancy.
- Systolic ejection click and an early systolic murmur in the second left intercostal space with transmission to the back. S2 may split widely.
- ECG evidence of right ventricular hypertrophy.
- Dilatation of main and left pulmonary arteries on chest radiograph.
- Right ventricular hypertrophy, systolic doming of the pulmonary valve, and a transpulmonic gradient by Doppler echocardiography.
Pulmonary valve, or pulmonic stenosis (PS), is the second most common form of congenital heart disease in the adult. Although many cases are so mild that they require no treatment, it often coexists with other congenital cardiac abnormalities (atrial septal defect [ASD], ventricular septal defect [VSD], patent ductus arteriosus, or tetralogy of Fallot [TOF]). Pulmonary valve stenosis is characterized by a conical or dome-shaped pliant valve with a narrow outlet at its apex. Right ventricular (RV) outflow is obstructed depending on the size of the orifice, and RV stroke volume may not rise appropriately during exercise. In response to the pressure overload, the RV hypertrophies, with an increase in wall thickness. This compensatory hypertrophy can involve the infundibulum and potentially lead to reversible dynamic subpulmonic stenosis once the valvular stenosis is relieved. If severe stenosis remains untreated, RV failure may ensue. It is important to differentiate pulmonary valve stenosis from stenoses of the peripheral pulmonary arteries and primary infundibular stenosis, often associated with VSD (see Tetralogy of Fallot). Pulmonary stenosis from a thickened, dysplastic valve is seen in patients with Noonan syndrome (a heterogeneous malformation syndrome with autosomal dominant inheritance).
The patient with PS usually has exercise intolerance in the form of exertional fatigue, dyspnea, chest pain, or syncope. RV failure with systemic venous congestion occurs late in the course of the disease. If the foramen ovale is patent or a concomitant ASD exists, shunting of blood from the right atrium to the left may occur, causing cyanosis and clubbing. The volume overload of pregnancy may precipitate right heart failure in patients with severe PS, although mild and even moderate stenosis are usually well tolerated.
In significant PS, the physical examination demonstrates a parasternal RV heave, a delayed and diminished or absent P2, and a late-peaking crescendo-decrescendo murmur that increases in volume with inspiration. If the valve is pliable, an ejection click precedes the murmur. This pulmonic ejection sound, best heard in the second left intercostal space, is the only right-sided event that decreases in intensity during inspiration and increases during expiration. As the stenosis becomes more severe, the systolic murmur will peak later in systole and the ejection click moves closer to the first heart sound, eventually becoming superimposed on it. The jugular venous pulse shows a prominent a wave, as a result of the diminished RV compliance, but jugular venous pressure is increased only in the late stages when RV failure occurs. Similarly, there may be an RV S4 gallop early in the course of the disease and a right-sided S3 in the later stages.
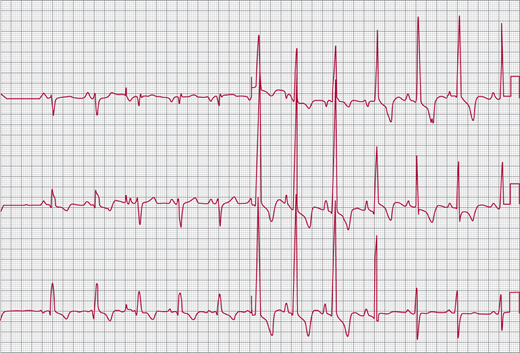
The ECG demonstrates evidence of right ventricular hypertrophy (RVH) with right-axis deviation, prominent R waves in the right precordial leads, and deep S waves in the left precordial leads (Figure 31–3). There may also be evidence of right atrial enlargement with peaked inferior (II, III, aVF) P waves.
The cardiac silhouette on the chest radiograph is normal in mild-to-moderate PS but may become enlarged in severe stenosis when right heart failure occurs. The main and left pulmonary arteries are often dilated. In addition to this “poststenotic dilatation,” dilatation may be seen even in cases of mild PS and may be related to intrinsic abnormalities of the pulmonary artery (idiopathic pulmonary artery dilatation).
The poor near-field resolution of transthoracic echocardiography (TTE) often limits definition of pulmonary valve morphology in the adult patient. When examined from the parasternal short-axis view, the valve may appear thickened (rarely calcified) and usually manifests systolic doming. In the absence of right heart failure, the RV dimension is normal or only mildly increased, but the RV wall thickness is increased (> 5 mm). In severe cases, the septum may be deviated toward the LV from the pressure overload of the RV. The right atrium and ventricle dilate late in the course of the disease. Saline contrast echocardiography should be performed in all patients with pulmonary valve stenosis to exclude an ASD or a patent foramen ovale.
Color-flow Doppler imaging demonstrates high-velocity flow within the pulmonary artery and is helpful in excluding a VSD or a patent ductus arteriosus. Continuous wave Doppler demonstrates a high-velocity jet across the RV outflow tract (Figure 31–4B). This signal is best obtained from the parasternal short-axis or subcostal short-axis views where flow is axial to the Doppler beam. PS is classified as mild when the RV systolic pressure is < 50 mm Hg, moderate when the gradient is 50–79 mm Hg, and severe when the gradient is > 80 mm Hg. Unfortunately, because of the lack of range resolution, continuous wave Doppler cannot localize the level of the obstruction. The morphology of the valve by echocardiography and pulsed wave Doppler mapping may provide localizing information, but additional diagnostic procedures are often necessary.
Figure 31–4.

A: Transesophageal echocardiographic views of a pregnant woman with severe pulmonary valve (PV) stenosis. The left image demonstrates the doming pulmonary valve in systole. The right frame illustrates the severe infundibular hypertrophy. B: Continuous wave Doppler recording from the same patient demonstrated a peak velocity of 6 m/s corresponding to a peak transvalvular gradient of 144 mm Hg.
TEE provides excellent definition of the RV outflow tract and pulmonary valve in the basal longitudinal views and excellent images of the atrial septum. As a result, noninvasive methods may now be adequate for establishing the diagnosis, even in adults.
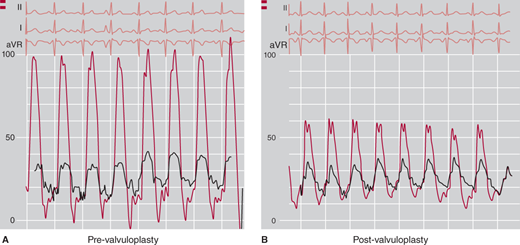
In most patients, cardiac catheterization is therapeutic as well as diagnostic because percutaneous balloon valvuloplasty has virtually replaced surgery for treatment of pulmonary valve stenosis (see below). During right-heart catheterization, the level of the stenosis can be confirmed by pressure monitoring during pullback from the pulmonary artery and supplemented by RV angiography. In valvular stenosis, there is a rise in peak systolic pressure as the catheter tip passes from the pulmonary artery into the infundibulum. In contrast, when the stenosis is in the infundibulum, the systolic pressure increases when the catheter is pulled into the body of the RV. As mentioned earlier, in PS, secondary hypertrophy may result in some degree of infundibular stenosis, and a pressure differential may be demonstrated at both levels on pullback (Figure 31–5). If the level of obstruction is still uncertain, cine-angiography may show the hypertrophied infundibulum or, alternatively, the domed and thickened pulmonary valve. Of course, both levels of obstruction may coexist.
Unlike aortic valve stenosis, the valve area is not calculated from the Doppler or invasive hemodynamic data, and the gradient alone is used to determine the severity of the obstruction and guide therapy.
In severe untreated pulmonary valve stenosis, the average life expectancy is approximately 30 years. The natural history of medically treated mild (gradient < 50 mm Hg) or moderate (gradient 50–79 mm Hg) PS and surgically treated severe (gradient > 80 mm Hg) PS is excellent with a 25-year survival rate of 95%. Surgical valvotomy via a pulmonary artery incision has been extremely effective in long-term relief of pulmonary valve obstruction. Although approximately 50% of patients have mild-to-moderate regurgitation following surgery, it is seldom of hemodynamic significance, and reoperation is rarely necessary.
In children treated conservatively for PS, the likelihood of eventually requiring surgery is dependent on the initial gradient: less than 25 mm Hg, 5%; 25–49 mm Hg, 20%; and 50–79 mm Hg, 76%. In the adult, the indication for treatment of pulmonary valve stenosis is a peak systolic gradient in excess of 50 mm Hg. When the gradient is between 40 and 50 mm Hg, the decision to treat is based on the presence of symptoms, the age of the patient, and the degree of RVH (by echocardiography or ECG). Echocardiography, before and after exercise, may be an important technique to assess RV function in the presence of an increased gradient.
As mentioned earlier, most patients (including adults) with pulmonary valve stenosis are currently treated with percutaneous balloon valvuloplasty. The Registry of the Valvuloplasty and Angioplasty of Congenital Anomalies has listed 35 patients over the age of 20, among them a 76-year-old man. No significant complications occurred in adult patients, and the gradient was reduced from approximately 70 mm Hg to 30 mm Hg, with about 50% of the residual gradient caused by infundibular hypertrophy. Ongoing assessment of these patients indicate sustained long-term relief of the pulmonary valve gradient with progressive infundibular remodeling causing further reduction in the outflow tract gradient over time. Recent technical improvements leading to the development of low-profile balloon have decreased the risk of pulmonary regurgitation after dilatation. Based on these results, percutaneous balloon valvuloplasty appears to be the treatment of choice in adults with pulmonary valve stenosis. Severe pulmonary valve insufficiency after either balloon or surgical valvotomy is uncommon, but patients should be evaluated every 5–10 years for this complication. Even when PS is associated with severe infundibular stenosis and tricuspid regurgitation, the long-term results from balloon valvuloplasty are excellent.
- A widely split S2 without respiratory variation (“fixed split”) and a midsystolic murmur are characteristic.
- RV conduction delay (“incomplete right bundle branch block”) with vertical QRS axis (ostium secundum ASD) and superior axis (ostium primum ASD) on ECG.
- Prominent pulmonary arteries and RV enlargement (decreased retrosternal air space) on chest radiograph. Increased pulmonary vascular markings.
- RV dilatation, increased pulmonary artery flow velocity, and left-to-right atrial shunt by contrast and Doppler echocardiography.
- Oxygen step-up within the right atrium; right-sided catheter can pass into the left atrium across the defect.
ASDs make up 10% of congenital heart disease cases in newborns and are regularly encountered as new diagnoses in adults. The defects vary in size from the smallest fenestrated ASD (a few millimeters) to the largest defect—the complete absence of the atrial septum, or common atrium. The most common interatrial communication is a patent foramen ovale that is anatomically and physiologically not classified as an ASD.
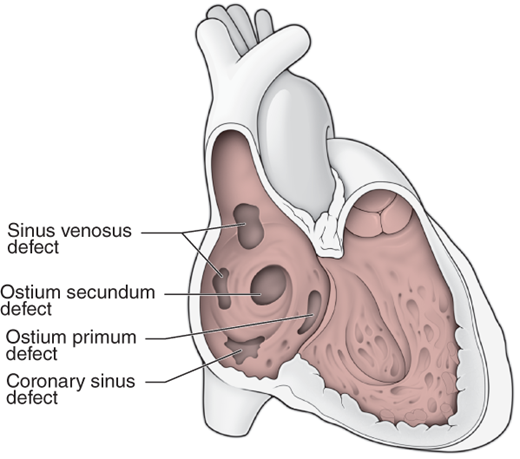
Classification of ASDs is according to location (Figure 31–6): ostium secundum in the region of the fossa ovalis, ostium primum in the lower portion of the atrial septum (actually part of an atrioventricular [AV] canal defect, discussed later), sinus venosus in the upper part of the septum near the entrance of the superior vena cava or at the entrance of the inferior vena cava, and unroofed coronary sinus (communication between the coronary sinus and left atrium). Important associated abnormalities include anomalous drainage of the right upper pulmonary vein into the superior vena cava associated with a superior sinus venosus ASD, a persistent left superior vena cava draining to the coronary sinus with secundum or primum ASDs, and a cleft anterior mitral leaflet and mitral regurgitation associated with an ostium primum ASD. Ostium primum ASD is a common cardiac anomaly in trisomy 21 (Down syndrome) and is part of the spectrum of AV septal “canal” defects (discussed later). There is an approximately 2:1 female predominance for ostium secundum ASDs, while the sex ratio for ostium primum and sinus venosus ASDs is approximately 1:1. An autosomal dominant inheritance pattern has been demonstrated in some patients with ostium secundum ASD with associated first-degree AV block, and cases of ASD in monozygotic twins have been reported. Recent studies have implicated mutations in the genes gata4, and nkx2-5 in nonsyndromic ASDs, whereas point mutations in the gene tbx5 are known to cause the Holt-Oram syndrome (ASD and limb defects).
The pathophysiologic consequences of an ASD depend on the quantity of blood shunted from the systemic to pulmonary circulation. The size of the shunt is in turn dependent on the size of the defect and the relative compliance of the RV and LV. Little or no shunting occurs immediately after birth because of the high pulmonary vascular resistance (PVR), but as resistance falls, the more compliant RV receives the shunted blood mainly in diastole, when all four chambers are in communication. In the compensated patient with ASD, pulmonary resistance is usually low. The older adult with the LV diastolic abnormalities of hypertension, coronary artery disease, and aging may experience increased left-to-right shunting and, consequently, right heart failure. While pulmonary resistance may increase, the development of Eisenmenger physiology is unusual after the age of 25. Atrial arrhythmias, especially atrial fibrillation, are common over the age of 50.
The young adult with an uncorrected ASD and normal pulmonary artery pressures is usually asymptomatic, with normal or minimally diminished exercise tolerance. After the age of 30, however, exertional dyspnea and atypical chest pain increase in frequency. As mentioned earlier, the frequency of atrial arrhythmias increases with age and occurs in a high percentage of patients over the age of 50 who have not been treated surgically. Signs and symptoms of RV failure may occur because of pulmonary hypertension or as a result of long-standing volume overload.
Important findings of the physical examination in an uncomplicated ASD include a prominent RV impulse along the lower left sternal border; a palpable pulmonary artery; a systolic ejection murmur, caused by increased flow across the pulmonic valve, which does not vary in intensity with respiration; and the almost pathognomonic fixed split second heart sound. When the Qp:Qs exceeds 1.5-2:1, there may be an associated right-sided diastolic flow rumble and S3 gallop from increased flow across the tricuspid valve. The patient with ostium primum ASD usually has a holosystolic murmur of mitral regurgitation. If pulmonary hypertension is present, P2 is increased and a high-pitched murmur of pulmonary regurgitation (Graham Steell murmur) may be audible. Signs of RV failure with elevated jugular venous pressure and venous congestion may be apparent in the later stages of this disease.
The ECG shows an RV conduction delay (“incomplete right bundle branch block” [IRBBB]) in 90% of cases (Figure 31–7). In ostium secundum and sinus venosus ASDs, the QRS axis is vertical or rightward. In the patient with ostium primum ASD, the axis is superior and leftward. Abnormal sinus node function in patients with sinus venosus ASD often results in an ectopic atrial rhythm with a superior P-wave axis.
The chest radiograph shows prominent main and branch pulmonary arteries with a small aortic knob and RV enlargement. The right atrium may appear enlarged. In the absence of pulmonary hypertension, the lung markings are increased as a result of increased pulmonary blood flow.
The findings on TTE include right-heart enlargement and increased pulmonary artery flow. Color-flow Doppler often can identify the interatrial flow, especially in the subcostal four-chamber view. An intravenous saline contrast injection should be used in all patients with these findings to exclude an unsuspected ASD. In the presence of an ASD, a negative contrast effect can be seen in the right atrium as the unopacified left atrial blood is shunted from left to right. A small degree of bidirectional shunting nearly always is present, and microbubbles can be seen in the left atrium as a result of right-to-left shunting. The shunting across a patent foramen ovale is purely right to left and occurs only during transient (eg, Valsalva maneuver, coughing) or persistent elevations in right atrial pressure.
Pulmonary artery pressure can be estimated from the peak velocity of the tricuspid regurgitant jet. Echocardiographic measurements may be used to determine shunt flow, eliminating the need for an invasive assessment. In adults, however, the TTE is somewhat limited in quantifying the magnitude of shunts and the size of the defect and in locating sinus venosus defects or anomalous pulmonary veins. As noted earlier, TEE has been found to be more accurate in determining the size and location of atrial communications (Figure 31–8). Biplanar and multiplanar transesophageal views are particularly useful in identifying sinus venosus type ASD (Figure 31–9).
Figure 31–8.
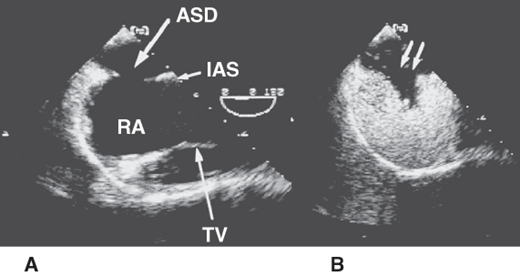
Transesophageal echocardiogram in a patient with an ostium secundum atrial septal defect (ASD). A: The image clearly demonstrates the position of the ASD in the midportion of the interatrial septum (IAS). B: The image is obtained after intravenous injection of agitated saline, which opacifies the right atrium (RA). The negative contrast effect produced by the unopacified left atrial blood entering the RA is clearly demonstrated (double arrow). TV, tricuspid valve.
Figure 31–9.
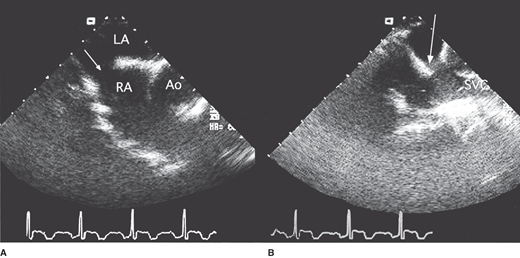
Transesophageal echocardiography in a 50-year-old man with a sinus venosus atrial septal defect. A: Horizontal view showing the defect (arrow) in the superior portion of the interatrial septum. B: The defect (arrow) is clearly demonstrated in this longitudinal plane view. Ao, aorta; LA, left atrium; RA, right atrium; SVC, superior vena cava.
In some younger individuals with unequivocally large defects on noninvasive imaging, diagnostic cardiac catheterization may be avoidable. In others, however, invasive studies may be necessary to accurately quantitate the shunt, measure PVR, and exclude coronary artery disease. Right-heart catheterization with repeated blood sampling for oxygen saturation demonstrates an oxygen step-up (ie, an increase in saturation) from the vena cava to the right atrium. In general, the higher the pulmonary arterial oxygen saturation, the greater is the shunt, with a value greater than 90% suggesting a large shunt. The ratio of pulmonary to systemic flow can be calculated by the following formula:
Where Sao2, Mvo2, Pvo2, and Pao2 are systemic arterial, mixed venous, pulmonary venous, and pulmonary arterial blood oxygen saturations, respectively. Mvo2 is calculated using the Flamm equation, [(3 × SVC) + IVC]/4, where SVC is the oxygen saturation of blood from the superior vena cava and IVC is the oxygen saturation of blood from the inferior vena cava.
A PVR that is more than 70% of the systemic vascular resistance suggests significant pulmonary vascular disease, and closure is best avoided. Pulmonary vasodilator therapy may be used, and occasionally, pulmonary resistance decreases enough to consider closure.
Although patients with an uncorrected ostium secundum ASD generally survive into adulthood, their life expectancy is not normal; older natural history studies showed a 50% survival beyond age 40. The mortality rate after the age of 40 is about 6% per year. Small ASDs (a Qp:Qs < 1.5–2:1) may cause problems only in the advanced years, when hypertension and coronary artery disease cause reduced LV compliance, resulting in increased left-to-right shunting, atrial arrhythmias, and potential biventricular failure. Severe pulmonary hypertension develops during young adulthood in only 5–10% of patients with large shunts (Qp:Qs > 2:1). Although most adults with ASDs have mild-to-moderate pulmonary hypertension, the late development of severe pulmonary hypertension in older adults appears to be quite rare. Pregnancy, in the absence of pulmonary hypertension, is usually uncomplicated. Another potential complication of ASD (including even the smallest patent foramen ovale) in the adult patient is paradoxical embolization. Endocarditis is rare in patients with ASD, and prophylaxis is not routinely recommended unless associated lesions with higher risk exist.
The natural history of sinus venosus ASDs is similar to that of ostium secundum defects, although many of these patients have associated partial anomalous pulmonary venous connection. Adults with an ostium primum ASD are less commonly encountered and may have additional complications resulting from mitral regurgitation caused by the cleft leaflet (see the discussion on AV canal defects, later in this chapter).
Ostium secundum ASDs have been surgically repaired for more than 40 years. No late cardiac deaths occurred in those who had early surgical repair of ASDs (before the age of 18) among patients in a large registry. Patients with elevated pulmonary systolic pressure (> 40 mm Hg) at the time of surgery have the poorest survival rate, especially if they are older than 40 at the time of operation.
Despite the poorer surgical results in adults older than 40 years, closure is superior to medical therapy and is recommended in patients with predominant left-to-right shunts (Qp:Qs > 1.5–2:1) and PVR less than 10 units/m2. Although mortality rates increase when the resistance exceeds this level, surgery can be performed safely in many patients with PVR between 10 and 15 units/m2; pulmonary vasodilator therapy should be considered in these patients before closure. Surgery will improve functional class and eliminate the risk of paradoxical embolization, but closure does not reduce the incidence of atrial arrhythmias.
Percutaneous device closure is widely available, and retrospective studies have suggested comparable results with device closure and surgical closure. Therefore, device closure has become the standard of care for appropriately selected adolescents and adults with ostium secundum defects.
Adult patients with initially small shunts (Qp:Qs < 1.5) should undergo continued echocardiographic surveillance because the shunt may increase over time owing to a progressive decline in LV compliance.
In patients with patent foramen ovale who have suffered embolic phenomena, device closure has become a standard intervention, although evidence from a randomized controlled trial is lacking.
- History of murmur appearing shortly after birth.
- Holosystolic murmur at left sternal border radiating rightward.
- Left atrial and LV or biventricular enlargement.
- High-velocity color-flow Doppler jet across VSD.
- Increased pulmonary flow velocities.
Because of the tendency for many VSDs to close spontaneously (see later discussion) and the tendency of larger defects to produce CHF in early childhood, it is relatively uncommon to encounter adults with previously undiagnosed VSDs of hemodynamic consequence. In adults, VSDs are usually either small and hemodynamically insignificant or large and associated with Eisenmenger syndrome. The importance of identifying the former is that they pose an ongoing risk of endocarditis and the potential complication of progressive aortic regurgitation. Eisenmenger syndrome is discussed later in this chapter.
Classifications of VSDs can be based on anatomic location or physiology. The anatomic classification includes defects of both the membranous and muscular portions of the ventricular septum (Figure 31–10). Membranous VSDs can be subdivided into supracristal (also known as doubly committed subarterial), perimembranous (the inlet portion of the membranous septum), and malalignment (found in TOF with an overriding aorta) defects. The muscular VSDs, often multiple, may be located in the inlet or outlet regions or within the trabecular portion of the septum. Classifying VSDs physiologically is based on the size of the defect as well as the relative vascular resistances within the systemic and pulmonary circulation. A high-pressure gradient exists across a small restrictive VSD, with normal or mildly elevated pulmonary artery pressure and predominant left-to-right shunting. A large nonrestrictive VSD permits equalization of RV and LV pressures with obligatory pulmonary hypertension (in the absence of RV outflow-tract obstruction) and bidirectional shunting. The smallest VSD (maladie de Roger) is characterized by a hemodynamically insignificant shunt, a loud murmur, and an intermediate to high risk of endocarditis.

In the infant, left-to-right shunting occurs only when PVR falls below systemic vascular resistance, and the murmur usually becomes audible in the first month of life. With a large nonrestrictive defect, PVR may not fall; if the defect is not surgically closed by age 2, irreversible pulmonary hypertension may ensue. The volume overload caused by a large restrictive VSD may cause CHF in the first 6 months of life. Approximately 40% of VSDs close spontaneously by age 3, and a smaller percentage close before age 10. Generally, the smaller defects are more likely to close, but even in infants with heart failure, 7% will experience spontaneous closure.
Three late complications of VSD are worth mentioning. Tricuspid regurgitation may rarely result when the septal leaflet of the tricuspid valve is deformed by the ventricular septal aneurysm that causes spontaneous closure of a perimembranous VSD. Aortic regurgitation is common in patients with doubly committed subarterial VSDs (supracristal, or outlet, VSDs), as a result of herniation of the right aortic sinus into the defect; it also occurs in those with perimembranous VSDs. Infundibular PS from hypertrophy of the RV outflow tract can develop, functionally dividing the RV into inflow and outflow segments, a condition termed “double-chambered right ventricle.” If a sufficient pressure gradient develops, RV systolic pressure can exceed LV systolic pressure, and right-to-left shunting can occur across the VSD. The resultant hypoxia may only occur during exercise.
The young adult with an uncorrected VSD and normal pulmonary artery pressures is usually asymptomatic, with normal or minimally diminished exercise tolerance. Like those with ASDs, exertional dyspnea often develops in patients with VSDs after the age of 30 when the Qp:Qs exceeds 2–3:1. Individuals with smaller shunts rarely report symptoms. The most disabled group with pulmonary hypertension and cyanosis (Eisenmenger physiology, or syndrome) will be discussed later.
Physical findings depend on the size of the VSD. The patient with uncomplicated VSD is acyanotic, and the LV apical impulse is displaced laterally, suggesting LV volume overload, and may be hyperdynamic. A holosystolic murmur occurs, often associated with a systolic thrill, heard best in the fourth or fifth intercostal space along the left sternal border, with radiation to the right parasternal region. Because of the increased flow across the mitral valve, an S3 gallop and a diastolic rumble may be present. Additional signs of tricuspid insufficiency (prominent jugular venous v wave and systolic murmur) or aortic valve regurgitation (diastolic blowing murmur, increased arterial pulses) will be present in patients with these complications.
In the presence of a large shunt, the ECG is suggestive of LVH or biventricular hypertrophy, with biphasic QRS complexes in the transitional precordial leads. Evidence of left or right atrial enlargement is present in only about 25% of patients.
Cardiac enlargement with an increased cardiac silhouette is evident on chest radiograph only in the presence of a large left-to-right shunt. In the absence of pulmonary hypertension, there is evidence of pulmonary vascular engorgement with a plethora of the peripheral vasculature as well as enlargement of the proximal vessels. Left atrial enlargement may be evident on the lateral chest radiograph.
It is important to remember that in most adults with a small VSD (< 1.5–2:1 shunt), both the ECG and radiograph are normal, even in the presence of a loud murmur. On the other hand, the presence of pulmonary hypertension alters the ECG and radiograph findings.
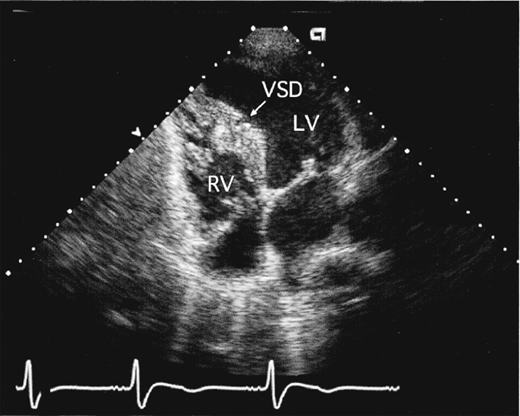
Two-dimensional and Doppler echocardiography can usually define the location and often the size of a VSD, although accurate Doppler shunt quantitation may not be possible in the adult. There is evidence of left atrial and LV dilatation. The right-heart chamber dimensions are usually normal, although the main pulmonary artery may appear dilated. The presence of RVH usually signifies pulmonary hypertension or associated PS (with right-to-left shunting and cyanosis). Usually only the largest defects, often located in the membranous septum, can actually be visualized echocardiographically (Figure 31–11). The aneurysmal pouch of a ventricular septal aneurysm may be seen in the parasternal short-axis view just below the aortic valve in the inlet portion of the septum near the septal leaflet of the tricuspid valve. Saline contrast administration shows a negative contrast effect within the RV, and a small degree of bidirectional shunting is sometimes present, with microbubbles appearing in the LV.
Color-flow Doppler imaging demonstrates a high-velocity (aliased) systolic jet across the ventricular septum into the RV. The location of the jet provides the best guide to the location of the defect. In the parasternal short-axis view, the jet from a membranous VSD may be seen in the region of the tricuspid valve (perimembranous) or toward the pulmonary artery (doubly committed subarterial, or supracristal). Muscular VSD jets are best seen in the apical or subcostal four-chamber views (Figure 31–12).
In continuous wave Doppler, the peak velocity of the jet across the ventricular septum provides the peak systolic LV-RV gradient (using the modified Bernoulli equation). Subtracting this gradient from the systolic blood pressure gives the peak RV systolic pressure. In the absence of a pressure gradient across the RV outflow tract—including the pulmonary valve (which should be carefully sought)—the RV systolic pressure is equivalent to the pulmonary artery systolic pressure. Additional Doppler evidence of the left-to-right shunt is found in the increased pulmonary artery flow velocity.
In the postrepair patient, the VSD patch may or may not be apparent, depending on the size of the original defect. Once endothelialized, the patch may not cause acoustic shadowing (or distal echo blockout). Color-flow Doppler may demonstrate patch leaks at the peripheral suture lines of the patch in a small percentage of patients. Spontaneous closure of a VSD involving juxtaposed tricuspid valve tissue may cause significant tricuspid regurgitation. Varying degrees of aortic regurgitation may be present and are most often associated with membranous or supracristal VSDs.
Although the diagnosis is often made noninvasively, the decision to close a VSD rests on accurate measurements of the shunt ratio and the level of PVR. Catheterization is therefore often necessary for therapeutic decision making.
Right-heart catheterization with sequential measurements of oxygen saturation reveals a step-up within the body of the RV. As with an ASD, the higher the RV oxygen saturation, the greater is the degree of shunting. For the calculation of Qp:Qs, the same formula is used as for ASD, except that the mixed venous blood sample is drawn from the right atrium. Pulmonary artery pressures and vascular resistance should be measured, and a gradient across the RV outflow tract, including the infundibulum and the pulmonary valve, must be excluded. Left ventriculography in the cranial left anterior oblique projection will reveal the location of the defect as contrast enters the RV.
As previously mentioned, adults with large, uncorrected VSDs are uncommonly encountered. With an uncorrected VSD, the overall 10-year survival rate after initial presentation is 75%. Survival is adversely affected by functional class greater than New York Heart Association class I, cardiomegaly, and elevated pulmonary artery pressure (> 50 mm Hg). As in patients with ASD, surgery is generally recommended when the magnitude of the systemic-to-pulmonary-shunt ratio exceeds 2:1. Other indications for surgery may include recurrent endocarditis and progressive aortic regurgitation.
In patients with small VSDs treated either conservatively or with surgery, outcomes are identical for patients with a Qp:Qs < 2.0, normal PVR, no LV volume overload or VSD-related aortic regurgitation, and no symptoms of exercise intolerance.
Surgery for closure of VSDs has been available for more than 50 years, and long-term follow-up data are available. Surgery prior to age 2—even in infants with a large VSD, high pulmonary blood flow, and preoperative pulmonary hypertension—almost always prevents the development of pulmonary vascular obstructive disease. In patients who underwent surgery during the 1960s and 1970s, there is an approximately 20% incidence of residual left-to-right shunt and a persistent risk of endocarditis. Ventricular arrhythmias and right bundle branch block (RBBB) are more common with a repair performed via right ventriculotomy (eg, muscular or subarterial VSD); when possible, the right atrium is the preferred approach. The risk of sudden death and complete heart block is low. Most patients who have VSDs repaired in childhood survive to lead normal adult lives.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


