Fig. 3.1
Median sternotomy approach for mitral valve repair. (a) The arterial cannula is in the ascending aorta along with a cannula for antegrade cardioplegia (white). The vena cavae have been cannulated directly. (b) Transseptal approach to the mitral valve by incising across the fossa ovalis (dashed line)
Typical congenital mitral stenosis is repaired by a combination of techniques dictated by the morphology of the valve. The repair may include commissurotomy, papillary muscle splitting or thinning, and resection of secondary papillary muscles and chordae. A supramitral ring is repaired by excising the entire ring with care taken not damage the mitral valve leaflets. Splitting of the single papillary muscle is the most common technique for repair of a parachute mitral valve. Finally, in patients with a hammock type mitral valve or mitral arcade, multiple fenestrations within the subvalvar apparatus may need to be created.
3.3.2.2 Mitral Insufficiency
Clinical presentation. Patients with mitral insufficiency present in a similar manner to patients with mitral stenosis, with signs and symptoms of congestive heart failure. However, patients with mitral insufficiency rarely present as neonates. Mitral insufficiency causes volume loading of the left ventricle and an increase in the left atrial pressures with left atrial enlargement as well. The pulmonary venous congestion predisposes patients to recurrent pulmonary infections [11].
On physical examination, the apical impulse may be hyperdynamic particularly when there is left ventricular enlargement from severe insufficiency. Auscultation reveals an S1 that is normal or diminished; the S2 is widely split. The murmur of mitral insufficiency is its hallmark. It is a holosystolic murmur heard best at the apex with transmission to the left axilla [11].
Indications for surgery. Symptomatic patients should undergo surgical repair. In a recent clinical series of 93 infants and children undergoing mitral valve repair, Stellin et al. reported 52% of the patients having mitral regurgitation. Thirty percent of the patients in this series had significant symptoms (dyspnea, failure to thrive, cardiogenic shock); the remaining patients had mild symptoms or were asymptomatic. The mean age at repair was 4.5 years [24]. This compares favorably to Carpentier’s early clinical series of pediatric patients with mitral insufficiency where the average age of patients at the time of surgery was 6.1 years. Brizard recommends surgical intervention on pediatric patients with severe regurgitation irrespective of symptoms, but cautions that the decision to proceed with surgery must be tempered against the likelihood of achieving a successful repair [32].
Functional classification of congenital mitral regurgitation. According to Carpentier’s functional classification of mitral valve disease, congenital mitral regurgitation can be classified in similar categories as congenital mitral stenosis. Patients with Type I regurgitation have normal leaflet motion. The valve leaks due to a lack of coaptation of the leaflet tissue. In Type II regurgitation, there is prolapse of the leaflet tissue. Essentially, the leaflets do not close on the same plane. In Type III regurgitation, the valve leaks because the motion of the leaflets is restricted. The subtypes are the same as those described for congenital mitral stenosis (Type III1 and Type III2) [17].
Patients with Type I regurgitation can have an isolated cleft of the anterior leaflet, annular dilatation, or a defect of leaflet tissue. Common anomalies seen with Type II regurgitation include elongated chordae tendineae or papillary muscles. Type III regurgitation may be associated with a parachute or hammock type valve. In patients with normal papillary muscles, there may be commissural fusion or short chordae that may lead to a combination of mitral regurgitation and stenosis [17].
Surgical repair of congenital mitral insufficiency—cardiopulmonary bypass: The techniques for cardiopulmonary bypass are the same as previously described for patients with congenital mitral stenosis.
Isolated cleft mitral valve is an anomaly primarily of the anterior or septal leaflet; however, clefts of the posterior leaflet have been described. Morphologically, there is a central separation of the leaflet tissue; the cleft can be wide and extend to the mitral annulus. The orientation of the cleft is towards the left ventricular outflow tract rather than the inlet septum as seen in patients with endocardial cushion defects (Fig. 3.2) [33].
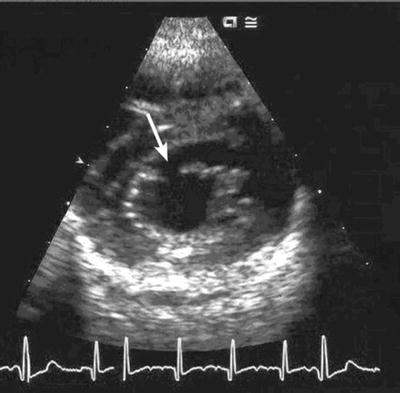
Fig. 3.2
Echocardiogram showing an isolated cleft in the anterior leaflet of the mitral valve (white arrow)
Cleft closure is achieved with multiple interrupted 7-0 polypropylene sutures. In the presence of annular dilation, the repair can be buttressed with a Kay-Wooler type annuloplasty or an annuloplasty band, depending on the age of the patient.
Patients with Type II regurgitation, based on leaflet prolapse and elongated chordae tendineae, can be repaired by chordal shortening. This surgical technique involves the creation of a groove within the body of the papillary muscle that supports the elongated chords. The excess length is buried within the papillary muscle, which is then closed (Fig. 3.3). Finally, the techniques for repair of a parachute mitral valve or hammock valve have been previously described.
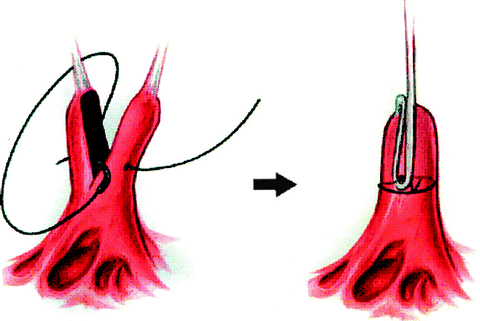
Fig. 3.3
Technique of chordal shortening. A groove is created in the body of the papillary muscle and the excess chordal length is buried within it (a). The groove is closed in (b)
3.3.3 Congenital Disorders of the Tricuspid Valve
Anatomy of the tricuspid valve. The tricuspid valve is the right atrioventricular valve; it has three leaflets designated the septal, anterosuperior (anterior), and inferior (mural) leaflet. The leaflet tissue is supported by a sub-valvar apparatus containing three papillary muscles which are extensions of the right ventricular wall and its extensive network of trabeculations. They are the inferior, medial, and anterior papillary muscles. The medial papillary muscle is also referred to as the muscle of Lancisi. One of the distinguishing features of the tricuspid valve is the presence of chordae that run from the free edge of the septal leaflet directly to the interventricular septum. This is a feature not seen with the mitral valve [16]. Also, the annulus of the tricuspid valve is about 20% larger than the mitral valve which allows ventricular filling at a low velocity and pressure [34].
3.3.3.1 Tricuspid Stenosis and Atresia
Congenital disorders of the tricuspid valve are considered primary valvar abnormalities when there is an intrinsic abnormality of the valve such as annular hypoplasia. This is in contrast to secondary tricuspid valve disease which is related to other disorders such as rheumatic heart disease, carcinoid heart disease, or other cardiac pathology such as right ventricular dysfunction. Secondary tricuspid valve disorders will not be discussed in this chapter [34]. Though rare, congenital tricuspid valve stenosis can be associated with right ventricular outflow tract obstruction, particularly pulmonary atresia with intact ventricular septum (PAIVS). This anomaly will be discussed in a separate section of this chapter.
The severity of isolated congenital tricuspid stenosis varies from mild annular hypoplasia associated with VSDs to complete absence of the valve (tricuspid atresia). Patients with mild annular hypoplasia rarely require intervention. On the other hand, patients with tricuspid atresia account for 1–3% of all patients with congenital heart disease [35].
Tricuspid Atresia
Clinical presentation. Tricuspid atresia is absence of the right atrioventricular valve and therefore absence of a direct connection between the right atrium and right ventricle. The primary consequence of this abnormality is a hypoplastic, rudimentary right ventricle. It represents one of the primary forms of univentricular congenital heart disease where the mature heart lacks two well-formed ventricles. With tricuspid atresia, a morphologic left ventricle is the driving force for systemic blood flow. The spectrum of univentricular congenital heart disease will not be considered in this chapter except as it relates to the overall objective [11].
The physiology of this lesion is characterized by an ASD that allows desaturated systemic venous blood to enter the left atrium, pass across the mitral valve, and fill the left ventricle to maintain systemic perfusion. An ASD is necessary for survival. In the presence of pulmonary stenosis or pulmonary atresia, pulmonary blood flow may be dependent on patency of the ductus arteriosus. The intracardiac shunt (ASD) causes severe systemic desaturation from birth. On physical exam, there is a single S2 with a systolic murmur at the lower left sternal border due to the presence of a VSD. When patients also have pulmonary valve stenosis, a palpable systolic thrill may be present [11].
Classification of tricuspid atresia. Patients with Type 1 tricuspid atresia have normally related great arteries, meaning the left ventricle gives rise to the aortic valve and the right ventricle gives rise to the pulmonary valve. This subtype represents 70% of patients with this malformation. Type 2 tricuspid atresia is associated with transposition of the great vessels, and Type 3 tricuspid atresia is associated with congenitally corrected transposition of the great vessels. A subset within each category is based on the status of pulmonary valve and the degree of pulmonary blood flow. In subtype a, there is pulmonary atresia. In subtype b, there is pulmonary stenosis or hypoplasia, and in subtype c the pulmonary valve is essentially normal. Furthermore, 30% of patients with tricuspid atresia have transposition of the great arteries and a VSD [36, 37].
Surgical management of tricuspid atresia (indications for surgery). The diagnosis of tricuspid atresia is an indication for surgical intervention. The overall objective for surgical management is to create a circulation where the desaturated systemic venous return bypasses the right atrium and is carried directly to the pulmonary arteries and lungs. This is accomplished with staged surgical procedures beginning in the neonatal period and usually concluding at 3–4 years of age.
Most patients with tricuspid atresia have inadequate pulmonary blood flow in the neonatal period, leading to hypoxia and poor arterial oxygen saturation. A stable source of blood flow to the lungs has to be created. A palliative aortopulmonary shunt can be created either as a direct communication between a systemic artery and the pulmonary artery, or a synthetic graft can be used. In 1945, Alfred Blalock, Chair of Surgery at John’s Hopkins University, and Helen Taussig, a pediatric cardiologist, were the first to describe the aortopulmonary shunt as a palliative procedure for the treatment of cyanotic congenital heart disease. The Blalock-Taussig shunt, as originally described, is a direct end-to-side anastomosis of the subclavian artery to the pulmonary artery [38, 39]. A common modification of the classic Blalock-Taussig shunt involves the interposition of a synthetic conduit (Gore-Tex) between the systemic artery and the pulmonary artery to create the communication between the two circulations (Fig. 3.4) [40].
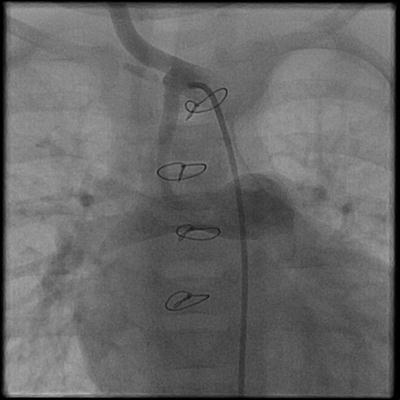
Fig. 3.4
Angiogram of a Gore-Tex-modified BT shunt between the innominate artery and the right pulmonary artery
Between 4 and 6 months of age, the “second stage” procedure in the series of operations to create a univentricular circulation can be performed. The Glenn shunt or bidirectional cavopulmonary connection is an end-to-side anastomosis of the superior vena cava to an undivided right pulmonary artery (Fig. 3.5). Consequently, the systemic venous return from the head and neck flows to both the right and left lung. As originally described by William Glenn at Yale University in 1958, the classic Glenn shunt was an end-to-side anastomosis of the right pulmonary artery to the superior vena cava. To make this connection, the right pulmonary artery is divided and sewn to the side of the SVC. Therefore, the systemic venous return from the head and neck flows to the right lung only [41].
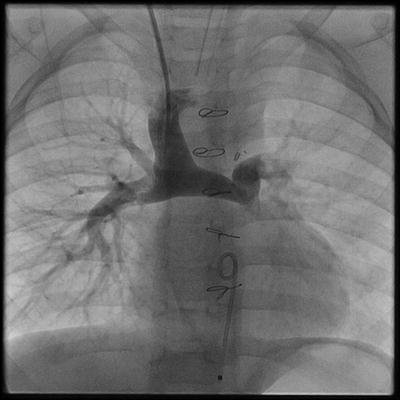
Fig. 3.5
Angiogram of a bidirectional cavopulmonary connection (BDG). The superior vena cava is anastomosed end-to-side to the undivided right pulmonary artery. In this arrangement, blood flows to the right and left lung
As stated, the overall objective of the surgical management of tricuspid atresia is to create a circulation where the systemic venous return bypasses the right atrium and flows directly to the lungs. As an intermediate step, the bidirectional cavopulmonary shunt achieves that goal for head and neck venous return. The “final stage” in this series of operations is to redirect the inferior vena cava blood directly to the lungs. Originally described by French cardiovascular surgeon Francis Fontan in 1968, the Fontan procedure is widely accepted as the definitive procedure in the creation of a univentricular circulation [42].
Since this original report, many modifications to the procedure have been made. These include the use of the modified Glenn shunt, staging of the Glenn procedure and the Fontan, absence of a valved conduit in the final corrective procedure, and the use of an extracardiac conduit to redirect the inferior vena cava return to the lungs [36]. Fontan and colleagues also suggested a set of selection criteria to improve outcomes with this procedure. This series of operations has become the definitive surgical pathway for the management of patients with single ventricle anomalies.
3.3.3.2 Ebstein’s Anomaly
Originally described in 1866 by Willhelm Ebstein, a German physician, Ebstein’s anomaly of the tricuspid valve is a rare congenital cardiac malformation, accounting for only 1% of all cases of congenital heart disease. The characteristic features of this anomaly are downward displacement of the septal leaflet and the mural leaflet towards the apex of the right ventricle, atrialization of a portion of the right ventricle, and abnormalities of the right ventricle. There may also be fenestrations of the leaflet tissue. The abnormalities of the leaflet tissue lead to poor coaptation with varying degrees of tricuspid regurgitation [43].
In addition to abnormalities of the leaflet tissue, the subvalvar apparatus is characterized by shortened chordae tendineae and papillary muscles. The right ventricle can be severely dilated, and the inlet segment is usually thinned or atrialized. However, the infundibulum or outlet segment of the right ventricle may be dilated as well. Consequently, there may be varying degrees of right ventricular dysfunction. VSDs, ASDs, and obstruction of the right ventricular outflow tract are other types of cardiac anomalies that may be associated with this congenital malformation [43].
Clinical presentation. The presence of tricuspid regurgitation, varying degrees of right ventricular dysfunction, and possible obstruction of the right ventricular outflow tract make the hemodynamic and clinical presentation of Ebstein’s anomaly variable. On the one hand, patients may be profoundly cyanotic in the neonatal period with ductal dependent pulmonary circulation. In these patients, an infusion of prostaglandin E1 (PGE) is used to maintain patency of the ductus arteriosus. On the other hand, patients with less severe forms of disease may be asymptomatic or only mildly symptomatic as neonates and present as older children and adolescents [11].
Neonatal Ebstein’s anomaly. Symptomatic neonates, with severe disease, present with cyanosis and possibly congestive heart failure soon after birth. When congestive heart failure is present, the child may be diaphoretic, tachypneic, have difficulty tolerating oral nutrition, and may tire easily. On physical exam, there is a systolic murmur of tricuspid regurgitation present at the lower left sternal border. The second heart sound is split as is S1, S3, and S4. The patient also has hepatomegaly, and older infants and children may have clubbing of the fingers and toes.
Right atrial enlargement, a right bundle branch block, first-degree AV block, and Wolf-Parkinson-White Syndrome may all be seen on the electrocardiogram. Massive dilation of the right atrium gives the classic chest X-ray appearance of a balloon-shaped heart with decreased pulmonary vascular markings. However, patients with mild forms of disease may have an essentially normal appearing chest X-ray [11].
Non-neonatal Ebstein’s anomaly. Even though patients with less severe forms of Ebstein’s anomaly may be asymptomatic as neonates, they need to be followed for the progression of tricuspid regurgitation and for the possible development of congestive heart failure. Also, right atrial enlargement related to worsening tricuspid valve regurgitation may cause intermittent episodes of supraventricular tachycardia. Patients who develop congestive heart failure can be medically managed with decongestive therapy including diuretics and possibly digoxin. Beta blockers can be used as prophylaxis for supraventricular tachycardia [11].
Surgical management of neonatal Ebstein’s anomaly. In 2009, Bove et al. from the pediatric cardiac surgery unit at the University of Michigan in Ann Arbor published their clinical strategy on the surgical management of neonatal Ebstein’s anomaly over 20 years (1988–2008). Of the 40 patients diagnosed with this cardiac malformation during that time period, 24 underwent surgery in the neonatal period; 16 patients were managed conservatively. The indications for surgery included symptomatic neonates who failed to wean from PGE, and the surgical strategy included a systemic-to-pulmonary-artery shunt alone in patients that were cyanotic after discontinuing PGE. Patients in whom the cyanosis improved but the heart failure symptoms persisted had either tricuspid valve closure and a systemic-to-pulmonary-artery shunt or a tricuspid valve repair alone [44].
Several techniques have been described for repairing the tricuspid valve in patients with Ebstein’s anomaly [45, 46]. Historically, the anatomic features of Ebstein’s anomaly that dictate the surgical repair are the dilated, atrialized portion of the right ventricle, which moves paradoxically during ventricular systole, and the presence of tricuspid regurgitation. One of the most widely applied techniques for tricuspid valve repair in patients with Ebstein’s anomaly was described by Danielson et al. from the Mayo Clinic. The fundamental components of the Danielson repair are as follows: plication of the atrialized portion of the right ventricle, an inferior annuloplasty of the tricuspid valve, and a reduction atrioplasty of the right atrium. This repair creates a competent unicuspid valve from the redundant anterior tricuspid valve leaflet [47]. Since their original report, several modifications have been made to the Danielson repair. These modifications include realignment of the anterior papillary muscle towards the ventricular septum and papillary muscle elevation which reportedly leads to enhanced coaptation of the leaflet tissue against the ventricular septum. Other techniques for tricuspid valve repair have been described by Carpentier which involve plication of the atrialized right ventricle and by Ullmann and Sebening who described a technique that does not involve exclusion of the dilated right ventricle [48, 49].
In 2007, da Silva et al. reported their experience with the so-called cone reconstruction of Ebstein’s anomaly. Based fundamentally on Carpentier’s techniques, the cone reconstruction involves detachment of the anterior and posterior leaflet as a single leaflet, clockwise rotation of this complex and reattachment to the true tricuspid valve annulus, plication of the tricuspid annulus, and valved closure of the ASD (Fig. 3.6). Da Silva et al. have used this technique in 40 patients with Ebstein’s anomaly from 1993 through 2005. There was one operative death (2.5%); two patients required tricuspid valve re-repair and no patients had complete heart block or required tricuspid valve replacement [46].
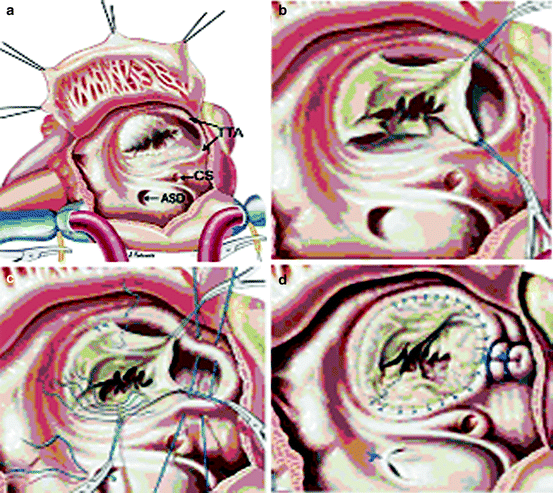
Fig. 3.6
(a) Cone reconstruction for Ebstein’s anomaly. (b) The anterior and posterior leaflets of the tricuspid valve are detached from the annulus as a single unit. (c) Clockwise rotation of the tricuspid valve leaflet tissue and plication of the dilated annulus. (d) Completed reconstruction Reprinted from the Journal of Thoracic and Cardiovascular Surgery, Volume 133, da Silva et al., “The cone reconstruction of the tricuspid valve in Ebstein’s anomaly. The operation: early and midterm results,” 215-23, 2007, with permission from Mosby, Inc
3.3.4 Straddling and Overriding Atrioventricular Valves
In 1979, Lev published an autopsy series of 96 hearts with straddling atrioventricular valves. In that series, straddling of an atrioventricular valve occurred when the tricuspid valve, mitral valve, or both crossed a VSD to lie in both ventricles. Two types of straddling were defined: basilar and peripheral. Basilar straddling occurred when the valve annulus crossed the VSD, while peripheral straddling involved only the subvalvar apparatus [50]. Basilar straddling has been redefined and is currently referred to as an overriding atrioventricular valve. So, an overriding atrioventricular valve is one in which the valve annulus is connected to both ventricles. The modern definition of a straddling atrioventricular valve is limited to the subvalvar apparatus, i.e., peripheral straddling as originally defined by Lev.
Several subtypes of straddling atrioventricular valves have been defined. Type A straddling occurs when the chordae tendineae cross the VSD and attach close to the defect in the opposite ventricle. Type B straddling occurs when the attachment point of the chordae tendineae is remote from the septal defect but remains on the septum. Type C straddling occurs when the chordae attach on the free wall of the opposite ventricle [51].
Straddling and overriding atrioventricular valves are extremely rare; they are seen in only 0.4–1.7% of most autopsy series examining congenital malformations of the heart. An overriding or straddling tricuspid valve is usually associated with a posterior VSD, and an overriding or straddling mitral valve is usually associated with an anterior VSD [52]. The associated anomalies dominate the clinical presentation. Other cardiac anomalies that may be associated with straddling or overriding atrioventricular valves include double outlet right ventricle, transposition of the great arteries, and Tetralogy of Fallot.
In patients with two well-formed ventricles, complete repair may be accomplished by VSD closure. Closure of the VSD may require making a slit in the VSD patch for passage of the anomalous chordae, resecting the chordae, division and resuspension of the chordae onto the VSD patch, or a univentricular pathway for patients with Type C straddling [53, 54].
3.4 Semilunar Valve Abnormalities
The function of the semilunar or arterial valves is to ensure unidirectional flow of blood from the ventricular chambers during systole. When blood is ejected from the ventricular chambers, the leaflets of the semilunar valves open. Closure of the semilunar valves occurs when the diastolic column of blood in the aorta and pulmonary artery exerts pressure on the valve leaflets [1].
3.4.1 Anatomy of the Semilunar Valves
3.4.1.1 The Aortic Root
When considering the anatomy of the aortic valve, it is useful to examine the anatomy of the aortic root complex. The aortic root is the junction between the left ventricular outflow tract and the ascending aorta. Its primary component is the aortic valve, but it also contains the sinuses of Valsalva, the membranous ventricular septum, and the coronary arteries. The aortic valve consists of three semilunar leaflets or cusps; each leaflet of the aortic valve is named for its respective coronary artery. Consequently, there is a right coronary cusp, a left coronary cusp, and a noncoronary cusp. The leaflets attach to the left ventricular outflow tract in a semilunar fashion creating triangular interleaflet spaces. At the apex of these triangles are the aortic valve commissures which are the highest points of attachment of the aortic valve leaflets onto the wall of the aorta. This junction, where the aortic valve commissures are hinged, is referred to as the sinotubular junction or ridge. It is a transition point from the aortic root to the ascending aorta [16].
Becker points out in a review of aortic valve functional anatomy that the sinotubular junction essentially is the outlet of the aortic root, and the lower attachments of the aortic valve leaflets to the left ventricular outflow tract represent the inlet to the aortic root. The ratio of inlet to outlet aortic root diameter is 1.34. The aortic valve leaflets, the sinotubular ridge, and the commissures are a functional unit. The noncoronary leaflet of the aortic valve is in fibrous continuity with the anterior leaflet of the aortic valve. In addition, the aortic valve also has fibrous continuity with the tricuspid valve by way of the membranous interventricular septum which is located beneath the junction of the noncoronary cusp and the left coronary cusp. The left ventricular outflow tract is both fibrous and muscular; the muscular component is the interventricular septum. The fibrous component is made up of the aorto-mitral curtain where the noncoronary cusp of the aortic valve is in fibrous continuity with the anterior leaflet of the mitral valve [55].
3.4.1.2 The Pulmonary Valve
Like the aortic valve, the pulmonary valve has three semilunar leaflets or cusps. The leaflets are named according to their relationship to the right and left coronary leaflets of the aortic valve; hence, there is a right and left facing leaflet. The third leaflet is referred to as the nonfacing leaflet. Unlike the left ventricular outflow tract which has a fibrous component in the anterior leaflet of the mitral valve, the right ventricular outflow tract is a complete sleeve of infundibular muscle [16].
3.4.2 Congenital Disorders of the Aortic Valve
3.4.2.1 Congenital Aortic Stenosis
Congenital aortic stenosis represents one of the more common forms of congenital heart disease accounting for 3–6% of all cases of congenital malformations of the heart [56]. The morphology of the aortic valve in this lesion varies. A bicuspid aortic valve is the most common congenital cardiac malformation with a prevalence ranging from 0.5 to 2% [57]. There are several morphologic subtypes of bicuspid aortic valves. In the most common type, there is fusion of the right coronary and left coronary leaflets (absence of right-left commissure). This subtype accounts for 70% of bicuspid aortic valves. Fusion of the left coronary and noncoronary leaflets is seen in approximately 1.4% of bicuspid aortic valves. Finally, there may be two essentially normal leaflets with no structural abnormality [58]. Other pathologic forms of congenital aortic stenosis include the unicuspid aortic valve with no discernable commissures and a tricuspid valve where the leaflets are fused and there is a central stenotic orifice [59].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


