Most surgical lung specimens from children are resections of cystic lesions and malformations, which are usually treated by excision of the abnormal lobe. The different cystic lesions and malformations often have overlapping radiographic features and clinical presentation, so the pathologist plays a key role in diagnosis and classification (Table 11.1).
The most commonly excised lung lesions from children are congenital pulmonary airway malformations (CPAMs). Other encountered malformations include bronchial atresia, intralobar and extralobar sequestrations, infantile lobar emphysema (congenital lobar overinflation), and bronchopulmonary foregut malformations. Bronchial atresia or some form of airway obstruction in utero may play a role in the pathogenesis of many of these malformations. The pathologic pattern may be determined by the timing of the obstructive event during lung development, as well as the level and completeness of obstruction.1,2,3
Clinical and Radiologic Findings
Children with congenital cysts and malformations of the lung are most often born at term. If the lesion is large, it may cause respiratory distress at birth due to compression or hypoplasia of the surrounding normal lung with subsequent respiratory compromise. Alternatively, if the surrounding lung is functioning normally, the child may be asymptomatic at birth and may present later in infancy or childhood with respiratory symptoms. Some lesions are asymptomatic and discovered incidentally, whereas others may present with symptoms and signs of pneumonia.
Radiographically, CPAMs, sequestrations, and lobar overinflation are usually limited to one lobe. They may appear as lucencies, densities, or cystic lesions on imaging. Sequestrations have a characteristic systemic vascular supply that is often apparent on imaging.
Congenital Pulmonary Airway Malformation
Terminology
CPAMs were formerly known as congenital cystic adenomatoid malformations (CCAM), but CPAM is now considered the preferred term for these lesions, since these lesions are “cystic” in only three of the five types and “adenomatoid” in only one type (type 3).
Incidence
As a group, they are the most common developmental lung malformation, with an incidence of about 1 in 5,000 live births.4
Localization
Over 90% of CPAMs are restricted to a single lobe; involvement of multiple lobes should alert the pathologist to other possibilities such as pleuropulmonary blastoma (PPB).
Clinical Findings
In a single-institution analysis of 172 cases of CPAM, the most common presenting symptoms were respiratory distress in infants under 6 months of age (40%) and recurrent pneumonia in older children (75%). Associated anomalies were seen in 47% of children, the most frequent being sequestration (71%) in association with type 2 CPAM. Mortality was 5%; risk factors for mortality were respiratory failure, sepsis, respiratory assistance requirements, and severe associated comorbidities.5
Classification
CPAMs have been classified into five major clinicopathologic types4,6 conceptually based on the putative anatomic level of the abnormality, that is, tracheobronchial to peripheral acinar. In a large series of CPAM, all histologic types were found; type 1 was the most frequent (70%), followed by type 2 (24%).5
Type 0 CPAM
The lesion historically described as type 0 CPAM is essentially synonymous with acinar dysplasia.7 These are extremely rare catastrophic developmental abnormalities that lead to profound pulmonary hypoplasia and are incompatible with life (see Chapter 12). This abnormality may be seen in term and premature infants and is associated with cardiovascular abnormalities and dermal hypoplasia. Grossly, the lungs are small and firm with a diffusely granular surface. Microscopically, there are no acini, and the lesion consists of bronchus-like structures with muscle, glands, and numerous cartilage plates.
Type 1 CPAM
Type 1 CPAM, the large or predominant cyst type, is the most common type of CPAM in most series. It presents primarily in the neonatal period but can be seen in older children and even adults. It is amenable to surgery and has a good prognosis. Grossly, type 1 CPAM is characterized by single or multiple large cysts (3 to 10 cm in diameter) surrounded by smaller cysts and compressed normal parenchyma.
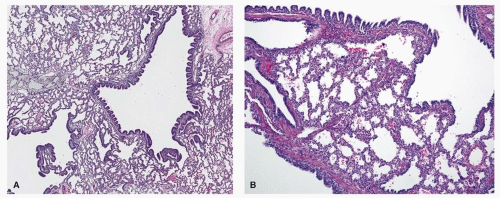
Microscopically, the cysts recapitulate proximal bronchi, being lined by ciliated, pseudostratified columnar epithelium with underlying fibromuscular tissue and focal cartilage plates. The epithelium usually shows a characteristic “sawtooth” architecture (Fig. 11.1). Almost half the cases show foci of mucous cells in the epithelial lining, which resemble benign gastric foveolar cells in most cases (Fig. 11.2). Identification of this mucigenic epithelium is strong evidence that the cystic lesion is a type 1 CPAM and thus can be useful in classification. The presence of mucigenic epithelium should always be reported if identified, since these mucous cells are recognized to be precursors for the rare examples of adenocarcinoma that have been described in association with type 1 CPAM.8
Type 2 CPAM
Type 2 CPAM conceptually recapitulates smaller bronchi and bronchioles, being composed therefore of medium-sized cysts. While historically thought to represent only 10% to 15% of cases of CPAM, it has likely been largely under recognized. Type 2 CPAM is a pattern observed in various other entities including bronchial atresia, intralobar sequestration, and extralobar sequestration. Therefore, recognition of the type 2 CPAM pattern should lead to careful consideration of these other entities, since some authors believe that essentially all cases of type 2 CPAM pattern are a result of intrauterine bronchial atresia or sequestration.
TABLE 11.1 Features of CPAM and Sequestration
Synonyms
Incidence, Associations
Clinical/Prognosis
Characteristic Features
Type 0 CPAM
Acinar dysplasia
Rare
Neonates, incompatible with life
Small, firm, retracted lungs
Type 1 CPAM
Large cyst type
Most common
Neonatal, older children, good prognosis
Sawtooth cysts, mucinous metaplasia
Type 2 CPAM
Medium cyst type
Associated with bronchial atresia, sequestration; 20% with extrapulmonary anomalies
Follows associated conditions, if present
Evenly distributed cysts that blend with surrounding lung; rhabdomyomatous dysplasia (occasional)
Type 3 CPAM
Small cyst/solid type
Rare
Neonatal, male predominance
Resembles immature lung
Type 4 CPAM
Peripheral acinar type
Rare; may be a form of regressed PPB
Newborns, young children
Thin-walled alveolar septa-like cyst walls; may appear similar to PPB, but without immature blastemal elements
Extralobar sequestration
—
Hydrops, polyhydramnios
CPAM type 2, pulmonary hypoplasia, diaphragmatic hernia
Uncommon
Prenatal, <3 mo; uncommon
Left hemithorax most common, then right hemithorax, mediastinum, and subdiaphragmatic; systemic feeder artery and venous drainage; outside normal pleural envelopment
Intralobar sequestration
—
Common
Childhood to adult
Systemic feeder artery; venous drainage pulmonary; lower lobes predominate, fibrosis; within normal pleural envelopment
PPB, pleuropulmonary blastoma.
About 20% of cases of type 2 CPAM pattern may have other associated congenital anomalies involving renal, cardiovascular, pulmonary, skeletal, and/or nervous systems,4,9 resulting in poor outcomes.
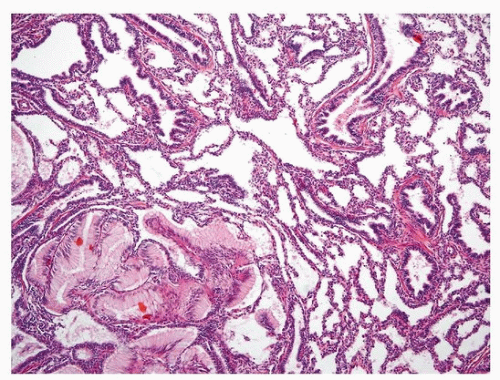
The type 2 lesion is composed of evenly distributed cysts measuring 0.5 to 2.0 cm in diameter that blend with the adjacent normal parenchyma (Fig. 11.3). Typically, there are bronchiole-like structures lined by cuboidal to columnar epithelial cells with a thin, underlying, fibromuscular layer (Fig. 11.4). These structures can be distinguished from normal bronchioles in that they lack a smooth muscle wall and lack an accompanying pulmonary artery branch. These bronchiole-like cystic structures may be back to back or may be more widely scattered with intervening alveolated parenchyma. Mucous cells and cartilage plates are absent except as components of “entrapped” normal bronchi.
FIGURE 11.1 ▲ Type 1 CPAM. A. There are cysts of various sizes, lined by a sawtooth pattern of bronchial epithelial cells. B. Higher magnification demonstrating cyst lining and adjacent alveolar spaces.
A subgroup of cases showing type 2 CPAM pattern will have so-called rhabdomyomatous dysplasia, characterized by ribbons of striated muscle fibers throughout the lesion, in association with the cysts, between alveolar ducts and around blood vessels (Fig. 11.5). Reported cases of associated rhabdomyosarcoma probably represent primary pleuropulmonary blastoma (PPB) rather than arising secondarily within type 2 CPAM.
Type 3 CPAM
Type 3 CPAM, the small cyst or solid type, is the originally described congenital cystic adenomatoid malformation.10 It is very rare (<5% of cases), usually presents in the first days to weeks of life, and has a male predominance. It tends to be large, is associated with maternal polyhydramnios and fetal anasarca, and has a high mortality rate. Grossly, it consists of a large, bulky, parenchymal mass involving an entire lobe or even an entire lung, with significant mass effect that may result in hypoplasia of the uninvolved lung. Cysts are typically uniform and <0.2 cm in diameter.
Microscopically, the lesion resembles an immature lung devoid of bronchi. There are irregular bronchiole-like structures lined with cuboidal epithelial cells surrounded by alveolar ducts and saccules that are also lined by cuboidal cells, imparting an “adenomatoid” appearance (Fig. 11.6). There is a paucity of blood vessels; mucous cells, cartilage, and rhabdomyomatous dysplasia are usually not present.
FIGURE 11.2 ▲ Type 1 CPAM. Foci of mucigenic epithelium (lower left) that gastric foveolar cells may arise in type 1 CPAM, and are thought to be the precursor to rare examples of mucinous adenocarcinoma that arise from CPAM.
Type 4 CPAM
Type 4 CPAM, the peripheral acinar cyst type, resembles the distal acinus. This variant may present in newborns or in young children (usually <5 years of age) with respiratory distress or pneumothorax, with some cases being detected incidentally.4,11 Imaging may show large air-filled cysts with mediastinal shift and pneumothorax. Grossly, large thin-walled collapsible cysts are present at the periphery of the lobe and appear to be lined by a smooth membrane (Fig. 11.7). Microscopically, the cysts are lined predominantly by flattened to cuboidal alveolar-type epithelial cells (Fig. 11.7), the underlying wall being composed of loose to fibrous mesenchyme with prominent arteries and arterioles.
Only gold members can continue reading. Log In or Register to continue