Chapter 63 Congenital Anomalies and Malformations of the Vasculature
Anomalous Venous Connections
Anomalous Pulmonary Venous Connections
Anomalous pulmonary venous connection refers to the absence of one or more pulmonary venous connections to the left atrium, without reference to the subsequent drainage of the anomalously disconnected pulmonary vein(s). Total anomalous pulmonary venous connection (TAPVC) refers to the bilateral absence of a connection of both pulmonary veins of each lung to the left atrium. The pulmonary veins connect directly into the right atrium or into one of its tributaries. TAPVC is almost always associated with some type of atrial septal defect (ASD) for life to be sustained beyond the newborn period.1Partial anomalous pulmonary venous connection (PAPVC) is the presence of connection of one or more—but not all—pulmonary veins to the right atrium or one of its tributaries. TAPVC is found in about 2% of autopsied cases with congenital heart disease and has a male predominance. PAPVCs constitute about 0.6% of autopsied cases of congenital heart disease.1 TAPVC is an isolated anomaly in two thirds of patients, and is associated with complex congenital heart disease in the remaining third, especially the heterotaxy syndromes (so-called isomeric TAPVC).
Embryology
In the human embryo, the lung buds and their pulmonary veins connect to the veins of the foregut, the splanchnic plexus. With growth, a new pulmonary venous channel, the primary pulmonary vein, grows as a bulging of the left atrium. At the same time, the intrapulmonary veins lose their connections with the splanchnic plexus and fuse with the primary pulmonary vein. The intrapulmonary veins eventually form the four pulmonary veins and are absorbed into the left atrium (Fig. 63-1). Failure of severance of the connection between the intrapulmonary and splanchnic veins results in anomalous pulmonary venous connections that may be total, partial, bilateral, or unilateral.
The drainage site of pulmonary veins may be supradiaphragmatic or infradiaphragmatic (Fig. 63-2). When drainage is supradiaphragmatic, both lungs may drain into a confluence, which may then drain into the left innominate vein and the superior vena cava (SVC), or the supradiaphragmatic drainage may be directly into the left SVC, or indirectly to the SVC via azygos or hemiazygos veins1 (see Fig. 63-2 ).
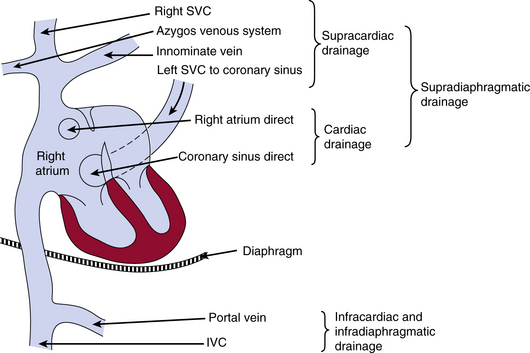
Figure 63-2 Possible sites of drainage for anomalous pulmonary venous connections into the venous system.
IVC, inferior vena cava; SVC, superior vena cava.
(Reproduced with permission from Becker AE, Anderson RH: Pathology of congenital heart disease, London, 1981, Butterworths, pp 47–66, 333.)
Maternal exposure to environmental teratogens such as lead paint or pesticides has been described to cause a familial susceptibility for TAPVC, largely in the presence of a positive family history of cardiac and noncardiac anomalies.2,3 Familial total anomalous pulmonary venous return is most likely an X-linked inheritance and autosomal dominant with variable expression and incomplete penetrance. Until now, two candidate genes have been proposed. The TAPVR1 gene, playing a role in vasculogenesis, maps to 4q12, in which the centromeric regions contain receptor tyrosine kinase genes as a kinase domain receptor (KDR). Recently it has been shown that dysregulation of the PDGFRA (platelet-derived growth factor receptor-α) gene causes PV inflow tract anomalies, including TAPVR.4
Total anomalous pulmonary venous connection
The level of the anomaly relative to the heart or diaphragm classifies TAPVC. Type I denotes anomalous connection at the supracardiac level; type II, anomalous connection at the cardiac level; type III, anomalous connection at the infracardiac level; and type IV, anomalous connection at two or more of those levels.5 The common forms of TAPVC are illustrated in Figure 63-3. In a recent multicenter study6 from the United Kingdom, Ireland, and Sweden with a cohort of 422 liveborn cases, the frequency of supracardiac TAPVC (type I) was 48.6%, infracardiac 26.1%, cardiac level 15.9%, and mixed connections 8.8%. Two cases (0.5%) had common pulmonary vein atresia, and 60 (14.2%) had associated cardiac anomalies. Of the supracardiac TAPVC cases, 73.2% showed connection to the innominate vein, 21% connected to various portions of the SVC, 2.9% connected to the azygos vein, and in 2.9% the connection was unknown (undocumented or unable to determine). The majority of cardiac-type TAPVC cases demonstrated connection to the coronary sinus (86.6%), followed by connections to the right atrium at 11.9%, and no identified connection site in 1.5% of cases. In the case of infracardiac TAPVC, the majority of cases demonstrated pulmonary venous connection to the portal system.6
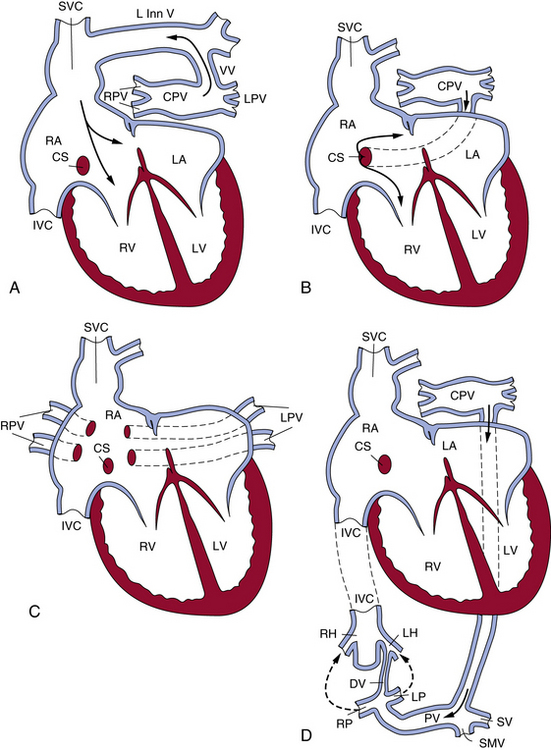
Figure 63-3 The most common patterns of circulation in total anomalous pulmonary connection (TAPVC).
(Reproduced with permission from Lucas RV, Krabill RA: Anomalous venous connections, pulmonary and systemic. In Adams FH, Emmanouilides GC, editors: Moss’ heart disease in infants, children and adolescents, ed 4, Baltimore, 1989, Williams & Wilkins, p 580.)
Size of the ASD has been shown to relate to longevity in TAPVC; the larger the ASD, the longer the survival. Individual pulmonary vein size at diagnosis also is a strong independent predictor of survival in patients with TAPVC.7
Associated cardiac anomalies include transposition of the great arteries, small ventricular septal defects (VSDs),6 pulmonary atresia, coarctation of the aorta (COA), and anomalies of systemic veins. There is a high frequency of TAPVC in patients with congenital heart disease and asplenia (heterotaxy syndromes). In a recent autopsy series of TAPVC associated with asplenia, anomalous pulmonary venous connection to a systemic vein was total in 42 (58%) of 72 and partial in 2 (3%) of 72, with obstruction in 24 (55%) of 44.8
The signs and symptoms, therefore, depend on the underlying hemodynamics—that is, presence or absence of pulmonary venous obstruction and the extent of mixing of blood between the right and left atrium. If interatrial mixing is inadequate, symptoms occur at birth or shortly thereafter. In TAPVC without pulmonary venous obstruction, patients are usually asymptomatic at birth, but at least 50% become symptomatic within 1 month of life (usually not in the first 12 hours of life). Once symptoms appear, however, they are rapidly progressive. Diagnosis may be established by angiography, echocardiography, or T1-weighted spin-echo magnetic resonance imaging (MRI).9
Partial anomalous pulmonary venous connection
The heart usually exhibits mild dilation and hypertrophy of the right atrium and ventricle, with dilation of the pulmonary artery. The left-sided chambers are normal. Usually, ASD accompanies PAPVC; conversely, 9% of cases of ASD have PAPVC. In the case of the right pulmonary vein connecting to the SVC, the right upper and middle lobe veins connect to the SVC. The vein of the right lower lobe usually enters the left atrium but may connect to the right atrium. The lower part of the SVC, below the azygos vein and above the right atrium, is dilated approximately twice the normal size. Most cases have associated ASD of the sinus venosus type, but occasionally a secundum or, rarely, a primum ASD may occur (Fig. 63-4).
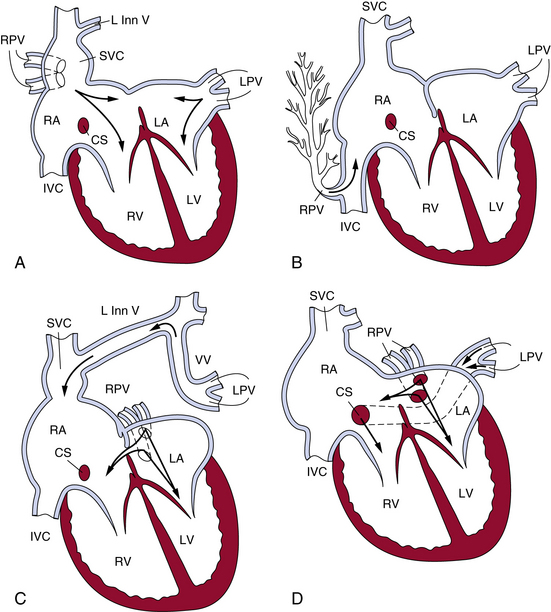
Figure 63-4 The most common patterns of circulation in partial anomalous pulmonary connection (PAPVC).
(Reproduced with permission from Lucas RV, Krabill RA: Anomalous venous connections, pulmonary and systemic. In Adams FH, Emmanouilides GC, editors: Moss’ heart disease in infants, children and adolescents, ed 4, Baltimore, 1989, Williams & Wilkins, p 580.)
Associated cardiovascular defects are common, occurring in up to 80% of patients.10 Major cardiac anomalies are found in about 20% of cases and include tetralogy of Fallot, VSD, single ventricle, COA, transposition of the great arteries, and hypoplastic left heart syndrome. Indications for surgery for isolated PAPVC include pulmonary-to-systemic flow ratio of over 2.0.10
When all right pulmonary veins drain the right middle and lower lobes and enter the IVC, either above or below the diaphragm, the pattern of the pulmonary veins is altered, giving a “fir tree” appearance (see Fig. 63-4). This malformation is called scimitar syndrome, as initially described by Chassinat in 1836,11,12 and derives its name from the shape of the anomalous vein on chest radiographs.13 Classically, this syndrome is associated with right lung anomalies, dextrocardia of the heart, hypoplasia of the right pulmonary artery, and anomalous connection between aorta and right lung. In the European Congenital Heart Surgeons Association (ECHSA) multicenter study, 65% of patients also presented with secundum-type ASDs, and 16% had a concomitant VSD.14
In connection of left pulmonary veins to the left innominate vein, the veins of the left upper lobe or the whole left lung connect to the left innominate vein via the vertical vein. Atrial septal defect of the secundum type usually is present, and the septum is rarely intact (see Fig. 63-4). Other sites of PAPVC are the left pulmonary veins draining into the coronary sinus, IVC, right SVC, right atrium, or left subclavian vein. In rare cases, veins of both lungs drain anomalously, but a small segment of the pulmonary venous system drains normally. That condition has been termed by Edwards subtotal anomalous pulmonary venous connection.
Cor Triatriatum
Cor triatriatum is a relatively rare cardiac anomaly (0.4% of autopsied cases with congenital heart disease, ratio of males to females 1.5:1).15 In this condition, the pulmonary veins enter an accessory chamber lying posterior to the left atrium and joining the left atrium through a narrow opening. The following broad classification has been suggested by a number of authors16,17:
1. Accessory atrial chamber that receives all pulmonary veins and communicates with the left atrium; no other connections (classic cor triatriatum).
2. Accessory atrial chamber that receives part of pulmonary veins and does not communicate with left atrium.
Physiologically, when blood from the accessory atrial chamber drains into the right atrium, the hemodynamic features are those of TAPVC. A stenotic opening between the accessory atrium and the left atrium, however, results in features of severe pulmonary obstruction. In subtotal cor triatriatum when obstruction affects only one lung, the result is reflex pulmonary arterial obstruction with decreased flow through that portion of the lung. The rest of the unobstructed lung receives increased flow, but there is no elevation in pulmonary arterial pressure (PAP).17
The classic cor triatriatum patients have onset of symptoms in the first few years of life. Patients usually have history of breathlessness and frequent respiratory infections. Right heart failure is usually present along with signs of pulmonary hypertension. Surgery has been advocated as early as the neonatal period,18 and balloon dilation may be successful in older children.19
Some patients remain asymptomatic until adulthood20 or may present with sick sinus syndrome in advanced years.21 In adults, clinical features on presentation can mimic those of mitral stenosis because of the obstructive properties of the membrane22; they include dyspnea with increased pulmonary capillary wedge pressure (PCWP) on exertion.23 As with other anomalies of the pulmonary venous system, associated anomalies may occur, including atrioventricular septal defect.24
Pulmonary vascular changes in cor triatriatum are progressive medial thickening and intimal fibrosis in pulmonary arteries and veins, accompanied by lymphangiectasia. In contrast to persistent left-to-right shunts, no plexiform lesions or more advanced stages of pulmonary vascular disease occur, which may explain the reversibility of PH due to congenital pulmonary venous obstruction.25
Congenital Stenosis of Pulmonary Veins
Congenital pulmonary vein stenosis is often associated with other anomalies, such as anomalous connections, cor triatriatum, and VSDs.26 The isolated form generally comes to clinical attention because of symptoms related to pulmonary hypertension.27 In its most severe form, congenital pulmonary vein stenosis is a progressive disease with rapid PH and rare survival beyond the first year of life.28
There are two types of stenosis of pulmonary veins. One is localized stenosis of the pulmonary veins at the junction of the left atrium, which may involve one or more pulmonary veins; the other is characterized by narrowing of the lumen of the pulmonary vein, either intra- or extrapulmonary in location, and is called hypoplasia of the pulmonary veins. The latter type is occasionally present in patients with pulmonary artery atresia or hypoplastic left heart syndrome. Clinically, patients have a history of respiratory symptoms, which may progress to right heart failure. Because of poor surgical results, balloon angioplasty and stenting currently are being evaluated as noninvasive treatments for pulmonary artery stenosis. In the most severe forms, lung transplantation appears to be an important option.28
Anomalous Systemic Venous Connections
Anomalous systemic venous connections, especially persistent left SVC, are usually asymptomatic and of little functional significance because systemic venous flow to the right atrium is not impaired. However, if they are not recognized, systemic venous anomalies may be accidentally severed during cardiac surgery. A classification based on embryological principles includes anomalies of the cardinal venous system, anomalies of the IVC, and anomalies of the valves of the sinus venosus.16
Anomalies of the cardinal venous system include persistent left SVC; persistent left SVC connecting to the right or left atrium, with and without failure of development of coronary sinus; right SVC connected to the left atrium; and anomalies of the coronary sinus. Anomalies of the IVC include double IVC, left IVC, and many lesser anomalies that are usually of no clinical significance, except that they may present problems to the surgeon.29,30 Infrahepatic interruption of the IVC with drainage via the enlarged azygous vein to the SVC has been found to occur in 2.9% of congenital heart defects.16 Anomalies of the valves of the sinus venosus involve the eustachian and thebesian valves and crista terminalis. Minor abnormal persistence results in larger valves and a Chiari network. Large outgrowth of the valve of the sinus venosus may result in complete or partial subdivision of the right atrium.
As with anomalous pulmonary veins, anomalous systemic venous return is a hallmark of the heterotaxy syndromes, especially asplenia. In a recent autopsy review of 72 cases of asplenia, the SVC was bilateral in 51 cases (71%) and unilateral in 21 (29%). In nine cases of bilateral SVC, one of the SVCs was partly or totally atretic. Although the IVC was never interrupted, a prominent azygos vein was found in six cases (8%). An intact coronary sinus was rare.8
Congenital Coronary Artery Anomalies
Anomalous Left Main Coronary Artery
The most common coronary anomaly resulting in clinical symptoms is an aberrant left main coronary artery arising in the right coronary sinus of Valsalva. There is a male/female ratio of 4:1 to 9:1. Sudden death occurs in up to two thirds of individuals with this anomaly, 75% of which occur during exercise.32 Most patients are adolescents or young adults, although death may occur as young as 1 month of age. There are often premonitory symptoms of syncope or chest pain, but stress electrocardiograms (ECGs) and stress echocardiograms are often negative.
Anomalous Right Coronary Artery
In contrast to anomalous left main coronary artery, anomalous right coronary artery from the left sinus is usually an incidental finding, although up to one third of patients may die suddenly. In the majority of cases (67%), the anomalous vessel courses between the aorta and pulmonary trunk, with the remainder usually coursing posterior to the aorta.32 Almost 50% of sudden deaths are exercise related, and most deaths occur in young and middle-aged adults (<35 years).
Anomalous Left Circumflex Artery
Anomalous left circumflex artery is the most common anomaly of the origin of a coronary artery, accounting for 28% of anomalies identified by cardiac catheterization and with an incidence of 1 in 300.33 This anomaly is considered benign. Awareness of this anomaly is important during cardiac surgery to avoid problems with myocardial protection or during prosthetic valve replacement.
Angiographic and Other Imaging in Diagnosis of Anomalous Coronary Arteries
Angiographically identifying an anomalous right or left main coronary artery from the contralateral coronary sinus depends on the vessel’s course between the aorta and pulmonary trunk. This particular course is angiographically distinct in the right anterior oblique projection when the left main forms a cranial-posterior loop (Fig. 63-5). An alternative method is to perform simultaneous pulmonary and coronary arteriography, or more practically, insert a pulmonary artery catheter as an angiographic marker for the location of the pulmonary vessels, and then perform an angiogram of the aberrant coronary artery in the steep anteroposterior caudal projection.
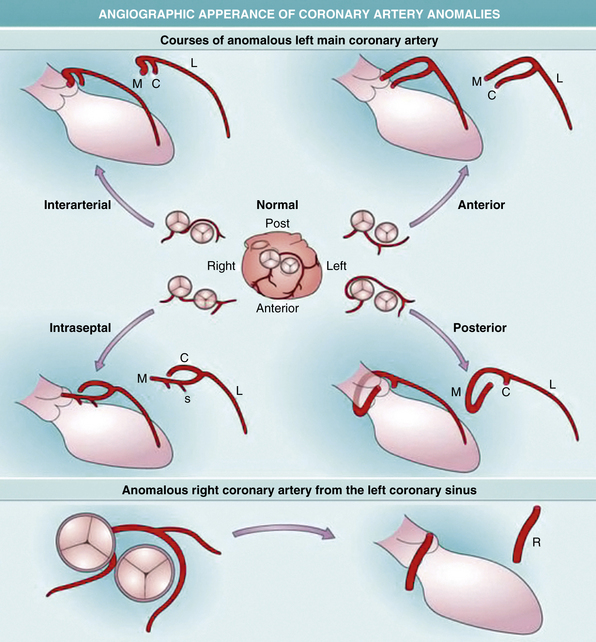
Figure 63-5 Development of coronary arteries.
(Reproduced with permission from Reese DE, Mikawa T, Bader DM: Development of the coronary vessel system. Circ Res 91:761–768, 2002.)
Standard two-dimensional (2D) echocardiography has been used as a screening procedure for coronary anomalies in athletes. An echocardiographic screening study of 3650 athletes found 3 cases of anomalous right (n = 2) and left (n = 1) coronary artery from the contralateral aortic sinus.34 However, the specificity of transthoracic echocardiography (TTE) is insufficient for this test to serve as an accurate screening tool.35 Alternative noninvasive procedures in the case of inadequate screening echocardiograms include transesophageal echocardiography (TEE) and the recent use of three-dimensional coronary magnetic resonance angiography (3D-CRMA). In one study, 3D-CRMA confirmed the anomalous origin of coronary arteries in 8 out of 15 (53%) pediatric cases, without the use of contrast medium or β-blockers, which could prove advantageous in the evaluation of younger patients.36
Malformations Affecting the Great Vessels
Coarctation of the Aorta
Coarctation of the aorta occurs as a congenital narrowing of the aortic arch, either as a discrete narrowing or one of some length that is usually located adjacent to the junction of the ductus arteriosus.37 The obstruction may be in the form of uniform tubular narrowing of some part of the aortic arch system, usually the isthmus (which lies between the left subclavian artery and the ductus arteriosus) or as a shelflike coarctation within the arch (Box 63-1). The latter is the more common of the two lesions. The narrowing may vary in severity but becomes significant only when there is a pressure gradient across the area of narrowing. That usually occurs when there is greater than 50% cross-sectional area reduction, but there may be lesser narrowing for tubular coarctation.
 Box 63-1 Classification of Coarctation of Aorta
Box 63-1 Classification of Coarctation of Aorta
Discrete Coarctation of Aorta (COA)
Modified from JE Edwards: Congenital malformations of the heart and great vessels. In Gould SE, editor: Pathology of the heart and blood vessels, ed 3, Springfield, Ill., 1968, Thomas, pp 391–454.
Incidence
Isolated COA is the fifth or sixth most common anomaly of all the congenital heart diseases, with estimates of 1 in 3000 to 4000 live births.38–41 In the New England regional study of congenital heart defects, COA accounted for 7.5% of anomalies in infants younger than 1 year of age.42 That may be an underestimation, since COA in newborn infants may not be detected because of similar blood pressure in the upper and lower extremities.43 The male-to-female ratio is 1.74:1.44 In older patients with isolated COA, the incidence is also higher in males. Most cases of COA appear to be sporadic, with no evidence of a mendelian pattern of inheritance.38 However, congenital heart disease has been reported in approximately 4% of the offspring of female COA patients.40 In addition, a recent nonparametric linkage analysis suggests a genetic basis for a subset of COA cases. McBride et al. demonstrated possible susceptibility loci on chromosomes 2p23, 10q21, and 16p12 in a cohort of 289 individuals from 43 separate families.40,45 Coarctation of the aorta is the most common cardiovascular defect found in Turner’s syndrome. Noncardiac abnormalities that have been reported with COA are hypospadias, clubfoot, and ocular defects.44
Pathology
The severity of luminal narrowing in the region of the coarctation at autopsy reveals 42% severe (pinhole) stenosis, 25% atresia, and 33% moderate narrowing in individuals 2 years of age and older.46 In cases of COA containing a contraductal shelf, there is an infolding of the aortic media into the lumen located opposite the ductus arteriosus, marked on the adventitial side by a localized indentation like a waist in the aortic wall.47 The aorta distal to the coarctation shows poststenotic dilation, and the wall of the aorta is thinner, whereas proximally where the pressure is higher, the wall is thicker. The shelf of the coarctation may be pre- or postductal, but most often is juxtaductal.48 Flow patterns in the fetus with various congenital defects determine the development of the aortic isthmus. In a normal fetus, the level of blood flow across the aortic isthmus is lower (25% of the combined total ventricular output) than after birth; therefore, the aortic isthmus at birth is narrower than the descending thoracic aorta.48 In congenital heart disease with reduced pulmonary arterial flow (pulmonary stenosis), the diameter of the isthmus is wider than normal because it carries greater flow in the fetus, and coarctation is virtually unknown. In lesions interfering with left ventricular (LV) outflow (mitral and aortic stenosis), the aortic isthmus is underdeveloped. When localized juxtaductal coarctation is present, aortic obstruction is not present during fetal life, but when the ductus arteriosus begins to close at birth, obstruction will appear after birth.49 The ductus closes from the pulmonary end; obstruction of the aortic end and a gradient across the coarctation may be delayed.
The ductal tissue itself plays an important role in the mechanism of coarctation formation. Normally, ductal tissue, composed mostly of smooth muscle cells (SMCs), extends only partially around the circumference of the aorta. In a patient with left-sided obstruction, there is right-to-left flow through the ductus in utero. This results in the migration of ductal tissue into the adjacent aortic wall, resulting in a circumferential distribution.48 Ho and Anderson, using a serial section technique, have confirmed that ductal tissue completely surrounds the juxtaductal aorta such that “the ductal and descending aorta form a common channel of structural continuity, and the isthmus enters this channel” rather than the ductus entering the isthmus descending aortic region.50,51
Collateral circulation
Olney and Stephens and Bahn et al. have reported that collateral circulation in infants who die with coarctation is poorly developed52,53 (Fig. 63-6). Bahn et al. noted that this depends on the location of the coarctation in relation to the ductus arteriosus; if the blood from the ductus enters the aorta proximal to the coarctation (preductal), then collateral circulation will develop53 (Fig. 63-7). If the ductal blood flow enters below the coarctation (postductal), however, there is little stimulation during fetal life for development of collateral circulation. In that situation, postnatal closure of the ductus results in obstructive hypertension and hypovolemia, which together result in LV failure.
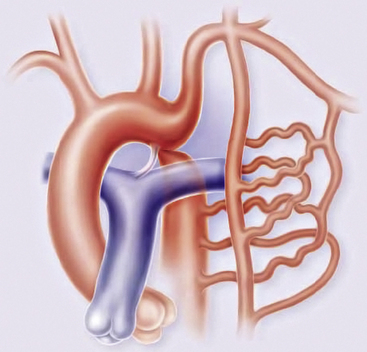
Figure 63-6 Coarctation of the aorta (COA).
(Reproduced with permission from Brickner ME: Congenital heart disease in adults. N Engl J Med 342:256–263, 2000.)
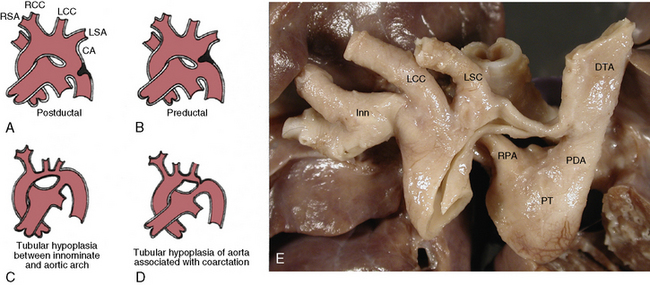
Figure 63-7 Coarctation of aorta (COA).
(A-D modified from Moller JH, Amplatz K, Edwards JE: Congenital heart disease, Kalamazoo, Mich., 1974, Upjohn, pp 46–51.)
The collateral circulation in COA between the proximal aorta and the distal aorta usually is present to some extent at birth but develops further as the patient ages. Collateral circulation primarily involves branches of both subclavian arteries, especially the internal mammary, vertebral, costocervical, and thyrocervical trunks, which carry blood to the lower limbs, usually through the third and fourth intercostal arteries and subscapular arteries. The subclavian arterial branches become greatly enlarged and are responsible for the classic signs of coarctation, such as rib notching, which extends from the third to the eighth rib, and parascapular pulsations.37,54,55 Collateral circulation also reaches the lower limbs through the internal mammary to the superior epigastric, which in turn connects with the inferior epigastric and joins the iliac arteries. The anterior spinal artery may provide additional collateral channels through its communication with the vertebral arteries above the coarctation and with intercostals and vertebral arteries below the coarctation.37,54
Development of good collateral circulation and clinical manifestations vary depending on the presence of stenosis of the left subclavian artery, which is an important source of collateral circulation; rib notching is seen only on the right side. If the right subclavian artery arises as a fourth branch and is distal to the coarctation, it is not a source of collateral flow, and rib notching occurs only on the left side.38
Associated conditions
Coarctation of the aorta is commonly associated with bicuspid aortic valve (BAV), but the exact incidence remains speculative (27%-46%).51,56 Among 250 patients with COA studied by Tawes et al.,56 bicuspid valve disease was present in 32 (13%). The most common lesion is stenosis and, unusually, incompetence, which occurs on the basis of bicuspid valve with persistent hypertension.57
There is a high incidence (85%) of major cardiac defects associated with COA in neonates48; and infants aged 1 to 11 months, however, the incidence drops to 52%. After 1 year of age, the incidence drops further to 40% (excluding bicuspid valve), as reported by Kirklin and Barratt-Boyes.57 Infants undergoing operation in the first 3 months of life have a 60% incidence of other congenital cardiac anomalies, compared to 25% of those between 6 and 12 months.57
Patent ductus arteriosus (PDA) is present in virtually all neonates and is considered part of the coarctation complex.57 Ventricular septal defect occurs in 30% to 36% of cases of COA.38,54 Coarctation of the aorta in transposition of the great arteries occurs in 10%, and ASD in 6% to 7%.58 Mitral valve disease, which causes mitral stenosis and regurgitation, is present in fewer than 10%.58
Complications
Aortic rupture usually occurs in the second or third decade and normally involves the ascending aorta, with resultant tamponade. Aortic rupture also may occur distal to the coarctation where the aorta is thin and dilated (poststenotic dilation).57 Some ruptures may be accompanied by aortic dissection.57 Women with coarctation are at high risk of aortic dissections during pregnancy. Stroke, as reported by Liberthson et al., usually occurs in older patients: older than age 40, 21%; age 11 to 39, 8%; and younger than age 11, less than 1%.58 The main cause is usually rupture of a congenital berry aneurysm in the circle of Willis and is secondary to the presence of hypertension.59
Aortic Arch Anomalies
Malformations of the aortic arch are a complex group of lesions first described in the 18th century. Classification of the aortic arch malformations is presented in Box 63-2. Vascular ring is the broad term used to describe an aortic arch malformation in which the trachea and esophagus are compressed. The first clinical description of tracheal and esophageal compression by a double aortic arch is credited to Wolman in 1939.60
 Box 63-2
Box 63-2Incidence
The true incidence of aortic arch malformations and vascular rings is difficult to determine because so many lesions are asymptomatic. Congenital heart defects involving the outflow tract, aortic arch, ductus arteriosus, and pulmonary arteries account for 20% to 30% of all congenital heart defects.61 The incidence of vascular rings is thought to be approximately 1% of congenital heart disease. Conotruncal malformations, including interrupted aortic arch and isolated anomalies of the aortic arch, are included in the constellation of findings in patients with the 22q11.2 deletion syndrome (del 22q11.2).62
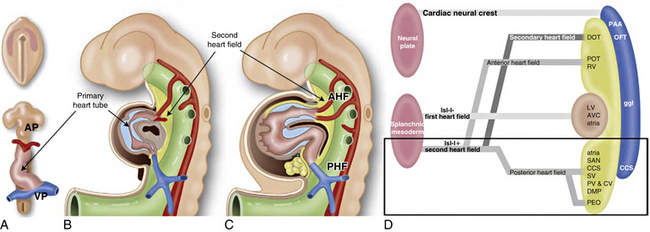
In 1948, Edwards proposed the hypothetical double aortic arch model to conceptualize all the known and possible aortic arch malformations.63 Today there is a better understanding of the embryology, and it has been shown that the final arrangement and morphology of the great arteries requires reciprocal signaling between endothelial cells (ECs) lining the pharyngeal aortic arteries and their surrounding SMCs and mesenchyme, derived from neural crest cells (NCC)64 (Fig. 63-8). Septation of the truncus into the aorta and pulmonary artery is accompanied by migration of the cardiac NCC into the pharyngeal arches and the heart, and this occurs in the mouse embryo at 9 (E9.0) days onwards. The NCC also contribute to the bilateral symmetrical aortic arches that arise from the aortic sac and undergo extensive remodeling, resulting in formation of the aorta, ductus arteriosus, and proximal subclavian, carotid, and pulmonary arteries. The NCC are remodeled to give rise to segments of the mature aortic arch, which is present from E12.5 days onwards.61 The left and right third arch arteries form the common carotid arteries (CCAs), the right fourth arch artery forms the proximal portion of the right subclavian artery, the left sixth arch artery forms the ductus arteriosus, and the right sixth arch artery regresses.65 Recent work with transgenic mice has shown which factors may be important for normal development of the arch vessels. For example, disruption of the forkhead transcription factor Mfh1 causes hypoplasia of the fourth aortic arch artery in mice, resulting in absence of the transverse aortic arch, resembling the interruption of the aortic arch in man.66
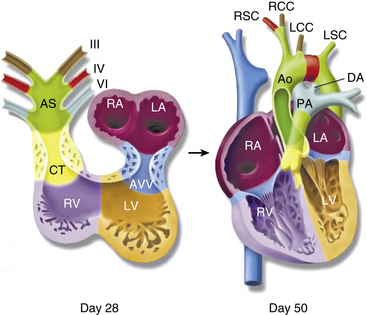
Figure 63-8 Schematic of development of arch vessels and outflow tract of the heart.
(Reproduced with permission from Srivastava D, Olson EN: A genetic blueprint for cardiac development. Nature 407:221–226, 2000.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


