The severity of obstruction varies considerably among patients. The smaller the orifice size of the obstruction, the greater is the level of systolic pressure required to eject the cardiac output through the obstruction. This principle is represented by the following equation:

During childhood, the heart usually maintains the elevated ventricular systolic pressure without dilation. Eventually, ventricular enlargement may appear because myocardial fibrosis develops. The fibrotic ventricular changes occur from an imbalance between the myocardial oxygen demands and supply. In most children, coronary arterial blood flow is normal, but with ventricular hypertrophy, myocardial oxygen requirements are increased.
Myocardial oxygen requirements are largely devoted to the development of myocardial tension and therefore are related directly to the level of ventricular systolic pressure and the number of times per minute the heart must develop that level of pressure. Thus, elevated ventricular systolic pressure and tachycardia increase myocardial oxygen consumption considerably.
With exercise, myocardial oxygen requirements increase even more in an obstructive lesion for two reasons: (1) cardiac output increases; so according to the relationship shown earlier, ventricular systolic pressure also increases; (2) with exercise, the heart rate increases.
If the increased myocardial oxygen requirements cannot be met, myocardial ischemia occurs and ultimately leads to myocardial fibrosis. These myocardial changes occur with time and lead to signs and symptoms. With the development of sufficient fibrosis, the contractile properties of the ventricle are affected so that ventricular dilation and cardiac enlargement develop.
As a group, the obstructive conditions are associated with normal pulmonary vascularity because the cardiac output is equal and normal on both sides of the heart and there is no shunting.
Children with obstructive lesions usually show few symptoms, but severe degrees of obstruction lead to congestive cardiac failure in neonates and young infants.
Coarctation of the aorta
Coarctation of the aorta (Figure 5.1) is a narrowing of the descending aorta that occurs opposite the site of the ductus arteriosus.
Figure 5.1 Coarctation of the aorta. (a) Central circulation before and after ductal closure; (b) repair options.
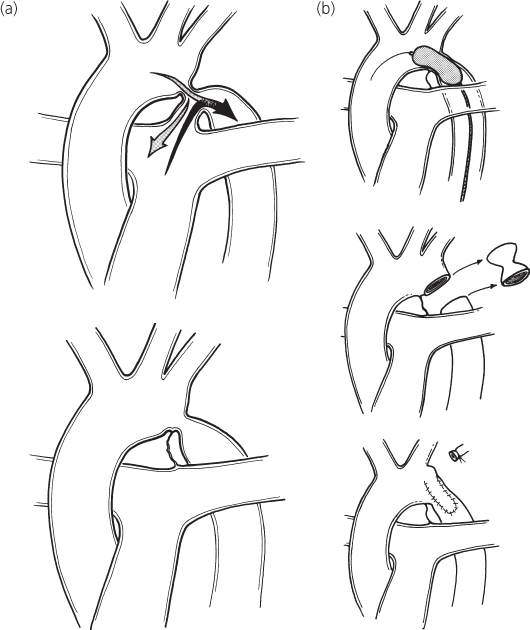
Aortic coarctation has traditionally been defined by its relationship to the ductus arteriosus, whether patent or ligamentous. This relationship has been described as either preductal or postductal. However, virtually all coarctations of the aorta are located juxtaductal (i.e. occurring in the wall of the aorta opposite the ductus arteriosus).
Coarctation may occur either as a localized constriction of the aorta or as tubular hypoplasia of the aortic arch and proximal descending aorta. In general, patients with tubular hypoplasia of the aortic arch develop cardiac failure in the neonatal period or early infancy. The coarctation in older children is usually discrete and is located distal to the origin of the left subclavian artery. Preoperative treatment and correction depend more on the associated lesions, such as arch hypoplasia, than on the precise relationship of the coarctation to the ductus.
The descending aorta beyond the coarctation usually shows poststenotic dilation. At least 50% of patients have a coexistent bicuspid aortic valve.
Coarctation of the aorta presents mechanical obstruction to left ventricular output. The pressure proximal to the coarctation is elevated, whereas that beyond the obstruction is either normal or lower than normal; this blood pressure difference is the major diagnostic feature of coarctation. In response to the pressure difference between the proximal and distal compartments of the aorta, collateral arteries develop between the high-pressure ascending and the low-pressure descending aorta.
Collateral vessels develop in any vascular system when a pressure difference exists. These vessels represent enlargement of naturally occurring small arteries bridging the high- and low-pressure components. Blood flows through these bridging vessels, and the volume of flow slowly increases, leading to the eventual dilation of the vessels. The internal mammary and intercostal arteries are the most frequently occurring collateral vessels in coarctation of the aorta.
Left ventricular hypertrophy develops in response to the elevated systolic pressure proximal to the coarctation.
History
Although most children with coarctation of the aorta are asymptomatic throughout childhood, 10% develop congestive cardiac failure during the neonatal period or early infancy. In the latter group, recognition of the lesion is important because proper management can be lifesaving.
Older children rarely develop congestive cardiac failure; instead, they have complaints, such as headaches, related to the systolic hypertension in the upper portion of the body. The very common childhood and adolescent symptom of chest pain, benign in most youngsters, occurs occasionally in coarctation patients and may be an ominous sign of myocardial ischemia secondary to severe left ventricular hypertrophy.
Coarctation of the aorta predominates in males at a ratio of 1.5:1. When coarctation of the aorta occurs in a female, Turner syndrome should be considered and chromosome analysis performed when appropriate. Some Turner syndrome patients exhibit very subtle findings and often escape clinical detection.
If coarctation of the aorta does not lead to congestive cardiac failure, the condition may be unrecognized until preschool age, when a murmur is heard, or later with the detection of hypertension.
Physical examination
Most patients show normal growth and development; many have an athletic physique. In neonates or infants, the signs of congestive cardiac failure may be present and profound. Mild degrees of acrocyanosis and mottling of the skin may be present because of pulmonary edema and poor perfusion, but these signs are common in healthy infants when cold.
Clinical diagnosis of coarctation of the aorta rests on identifying a blood pressure difference between the upper and lower extremities. This information may be gathered by palpation of both the radial and femoral arteries. If a substantial difference between the two is found, coarctation of the aorta should be suspected.
In addition, finding very sharp and brisk radial pulses in infants should lead one to consider coarctation of the aorta; radial pulses are ordinarily difficult to palpate in this age group.
Regardless of whether the femoral pulses feel diminished or not, the blood pressure should be taken in both arms and a leg in every child with a murmur. Coarctation of the aorta has been missed in many patients because the “femoral arteries were palpable.”
The blood pressure should be obtained either by direct auscultatory means or with automated devices (see Chapter 1). Blood pressure cuffs of appropriate width must be used. The largest cuff that fits the extremity should be used. In a patient without cardiac disease, the blood pressure should be the same in the upper and lower extremities. If the blood pressure is higher in the arms than in the legs by 20 mmHg or more, the difference is considered significant and indicates coarctation of the aorta. Using an inadequate-sized leg cuff can artifactually increase the leg pressures and lead to failure to detect a significant systolic pressure difference when one exists.
In infants with congestive cardiac failure secondary to severe coarctation of the aorta, the blood pressure values may be similar in the arms and legs, but at low levels at both sites, because cardiac output is so reduced. Following stabilization of such infants however, the pressure difference between the upper and lower extremities usually becomes apparent.
An open ductus, either native or from prostaglandin administration, palliates a neonate with coarctation and equalizes upper- and lower-extremity pulses because the aortic end of the ductus provides a bypass around the obstruction.
The examination of the heart may reveal cardiac enlargement. Palpation in the suprasternal notch reveals a prominent aortic pulsation and perhaps a thrill in patients with a coexistent bicuspid aortic valve. An ejection-type murmur is present along the sternal border, at the apex, and over the back between the left scapulae and the spine in the fourth interspace. The murmur is generally grade 2/6–3/6.
An aortic systolic ejection click is often heard, indicating dilation of the ascending aorta from a coexistent bicuspid aortic valve. The loudness of the aortic component of the second heart sound may be increased. In an infant with congestive cardiac failure, auscultatory findings may be muffled until cardiac performance is improved.
Electrocardiogram
The electrocardiographic findings vary with the age of the patient.
Neonate
In the neonatal and early infancy periods, the electrocardiogram usually reveals right ventricular hypertrophy.
Several explanations have been offered for this seemingly paradoxical finding. If the ductus arteriosus remains patent, the right ventricle, because of its communication through the pulmonary artery and ductus arteriosus, continues to work against the resistance imposed by the systemic circulation. In other patients with coarctation of the aorta and patent ductus arteriosus, the left ventricle is hypoplastic, so the electrocardiogram shows a pattern of right ventricular hypertrophy. Right ventricular hypertrophy has also been explained by the development of pulmonary hypertension secondary to left ventricular failure. The load placed on the fetal right ventricle, normally about 60% of the combined output from both fetal ventricles, may increase because less blood is able to traverse the left ventricle and to pass through the narrowed aortic isthmus.
Subsequently, the electrocardiogram shifts to a pattern of left ventricular hypertrophy.
Older infants
In older infants with severe coarctation of the aorta or in those with coexistent aortic outflow obstruction and endocardial fibroelastosis (representing subendocardial scarring) of the left ventricle, a pattern of left ventricular hypertrophy, inverted T waves, and ST depression in the left precordial leads is present. These ventricular repolarization abnormalities are often signs of a poor prognosis.
Older patients
In the older patient with coarctation of the aorta, the precordial leads show either left ventricular hypertrophy or a normal pattern.
Chest X-ray
In symptomatic infants, significant cardiac enlargement is present, with the cardiomegaly consisting primarily of left ventricular and left atrial enlargement. The lung fields show a diffuse reticular pattern of pulmonary edema and pulmonary venous congestion.
In older children, cardiac size and pulmonary vasculature are usually normal.
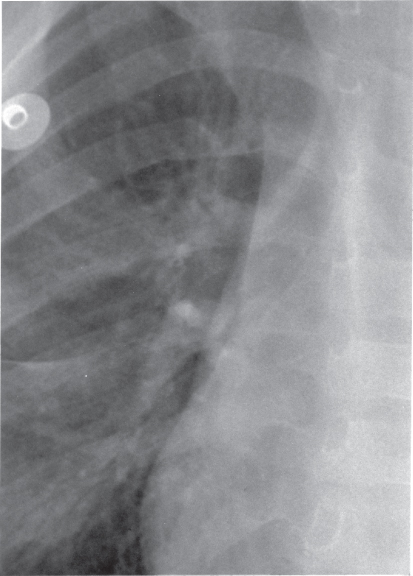
The appearance of the descending aorta is often diagnostic of coarctation of the aorta by showing poststenotic dilation. The barium swallow shows an E sign. The upper portion of the E is formed by the segment of the aorta proximal to the coarctation, and the lower portion of the E is formed by the deviation of the barium from the poststenotic dilation. On plain chest X-rays, often the left side of the thoracic aorta shows soft-tissue densities in the form of the number 3 that mirror the barium sign. The upper portion of the 3 sign represents the aortic knob, and the lower portion represents the poststenotic dilation. These findings help in identifying the extent of the coarctation.
The ascending aorta may be prominent with coexistant bicuspid aortic valve.
Rib notching (Figure 5.2) may be apparent in older children and adolescents, but its absence does not rule out the diagnosis of coarctation. The inferior margins of the upper ribs show scalloping caused by pressure from enlarged and tortuous intercostal arteries serving as collaterals.
Echocardiogram
Cross-sectional images of the aortic arch, usually best obtained with the transducer positioned near the suprasternal notch, reveal narrowing at the site of coarctation. In some patients, hypoplasia of the transverse segment of the aortic arch extends to the coarctation. The proximal thoracic descending aorta just distal to the coarctation may be normal in size or may be slightly dilated, representing poststenotic dilation.
Color Doppler shows a disturbed (turbulent) signal at the stenosis, and spectral Doppler shows high-velocity flow from the transverse aortic arch to the descending aorta with a continuous pattern (extending from systole into diastole).
In neonates, the diagnosis may be difficult as long as the ductus arteriosus remains large. Flow through a neonatal ductus in coarctation is bidirectional, often predominantly right to left (pulmonary artery to aorta). This is an important echocardiographic clue to the diagnosis.
The echocardiogram provides rapid assessment of left ventricular hypertrophy, size, and function and also allows diagnosis of possible associated lesions, such as bicuspid aortic valve, mitral valve malformations, and ventricular septal defect.
Cardiac catheterization and angiography
Usually, the clinical findings and echocardiogram are sufficient to diagnose coarctation of the aorta. Diagnostic catheterization and angiography are unnecessary, unless performed in conjunction with balloon dilation.
Oximetry data are usually normal, except in neonates with a large ductus. Pressure measurements demonstrate systolic hypertension proximal to the coarctation and a gradient at the site of the coarctation, often dramatically shown by pullback of the catheter across the lesion during pressure recording.
Treatment
Medical management prior to gradient relief
Infants with a coarctation of the aorta who develop congestive cardiac failure usually respond to medical management within a few hours and then undergo successful repair. Infants who fail to respond promptly to medical management or to reopening of the ductus with prostaglandin may require emergency repair. The operative risk is higher in these patients.
Assessment in preparation for gradient relief
To make appropriate operative decisions, the exact location of the coarctation of the aorta must be known. This is done by integrating information from the physical examination, and by directly imaging the lesion by echocardiography, angiography, MRI/MRA, or CTA.
The distal extent of the coarctation can be recognized by the identification of poststenotic dilation, and the proximal extent, by the blood pressure in the two arms. Usually, the recordings are similar in both arms, indicating that the coarctation is located distal to the left subclavian artery. Occasionally, the blood pressure of the left arm is lower than that of the right arm, indicating that the coarctation of the aorta involves the origin of the left subclavian artery and therefore a longer segment of the aorta.
Echocardiographic images are often limited by the size of older patients. MRI/MRA and CTA are particularly useful imaging techniques in adolescents and adults with coarctation, since the coarcted segment of the aorta has little motion throughout the cardiac cycle.
In an infant in cardiac failure, the diagnosis may be difficult, in which case aortography, MRI/MRA, or CTA may be helpful.
Surgery
Two major types of operation for coarctation are widely used.
Excision and end-to-end anastomosis
A discrete coarctation is excised and the two ends of the aorta are reanastomosed. An elliptical incision is made to minimize narrowing that may accompany growth of the patient and/or shrinkage of the anastomotic scar.
Subclavian flap repair
In patients with a very hypoplastic aorta or long-segment stenosis, the repair site can be augmented by transecting the left subclavian artery distally and opening it linearly to create a flap of living tissue. Early attempts to augment the arch repair with synthetic or pericardial patch material often led to late aneurysm formation.
Although long-term surgical results are very good, no operative technique is free from the risk of late restenosis.
Operation should be performed on most patients with coarctation of the aorta when the defect is diagnosed, except perhaps in a small, premature infant who can be palliated with prostaglandin infusion and allowed to grow to near-term weight. Doing this improves the efficacy of repair and minimizes the risk of late restenosis. The operative mortality risk is low (less than 1 in 400) in patients with an uncomplicated coarctation.
Infants with severe associated anomalies, such as a very large ventricular septal defect, small left ventricular outflow tract, and associated left ventricular failure from volume and pressure overload, may benefit from a staged repair. Repair of the coarctation and pulmonary artery banding first often leads to rapid improvement in the left ventricular dysfunction and eventual growth of the outflow tract. Several weeks or months later, removal of the band and closing of the ventricular septal defect follow. The operative mortality for one-stage neonatal repair of such infants can be higher than that of a staged approach.
Interventional catheterization
Balloon dilation of coarctation at the time of cardiac catheterization has been successful for native (previously unoperated) coarctation and for postoperative restenosis.
In postoperative restenosis, the results of gradient relief are good and the risk of balloon dilation is low, possibly due to the external buttressing of the dilated region by the old operative scar. Reoperation for restenosis carries increased risk compared with balloon dilation, partly because of the operative scarring, which must be dissected to achieve exposure.
Balloon dilation of native coarctation avoids some operative disadvantages but, compared with operative repair, it involves a greater chance of immediate complications such as extravasation and of late complications of aneurysm formation or restenosis. The age and size of the patient at the time of balloon dilation influence the risks and long-term outcomes: younger and smaller patients have higher risk.
Implantation of a metallic stent at the time of balloon dilation may lessen the risk of aneurysm formation but in small patients the stents do not allow for growth, hence repeat balloon dilation of the stented region is usually needed.
Natural history
The anastomotic site following coarctation repair may not grow in proportion to aortic diameter growth. Therefore, recoarctation may develop, often necessitating a second operation when the patient is older. This need occurs more frequently among children with a very hypoplastic aorta who were operated upon in infancy. Follow-up of all operated patients includes periodic determination of blood pressure in both the upper and lower extremities.
Since half of the patients with coarctation of the aorta have a bicuspid aortic valve, they are at some increased risk for development of endocarditis compared with persons with a normal aortic valve; however, antibiotic prophylaxis is no longer advised for most patients (see Chapter 12). The long-term course of patients with bicuspid aortic valve is variable as the valve may become slowly regurgitant or stenotic with age, and eventually require valvar surgery.
Following operation, some patients have persistent hypertension in both the arms and the legs. The reasons are not well understood, but it does not seem to be related to elevated levels of renin and angiotensin. Abnormal vascular reactivity has been demonstrated in patients with well-repaired coarctation. After repair, some patients with normal resting blood pressure have an exaggerated hypertensive response to exercise. This hypertension requires management. Delay in diagnosis and corrective surgery until an older age in childhood increases the risk of permanent systemic hypertension.
Aortic stenosis
Aortic stenosis can occur at one of three anatomic locations (Figure 5.3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


