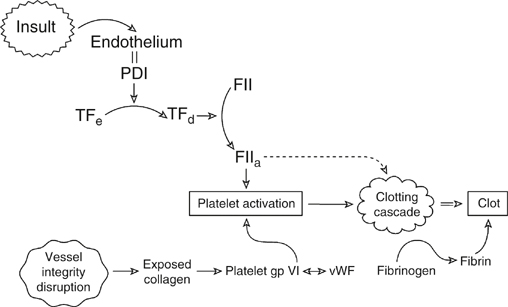
Hemostasis is generally initiated by damage to the vessel wall and disruption of the endothelium, but in venous thrombosis (VT), it may be initiated in the absence of vessel wall damage. Both arterial thrombosis and VT converge on platelets, tissue factor (TF), and thrombin (Figure 1). However, thrombosis in the arterial system occurs somewhat differently than in the venous system. The elements required for initiating VT were described by Virchow as stasis, endothelial injury, and hypercoagulability of the blood. In the arterial circulation, endothelial injury (whether acute or chronic) is key to thrombosis. This is most clearly demonstrated by the typical atherosclerotic plaque. In advanced lesions, the lipid core of the plaque is rich in inflammatory cells, cholesterol crystals, and TF (generated by activated macrophages within the plaque). Plaque ulceration exposes highly thrombogenic lipid to the blood stream, activating coagulation and platelet aggregation and leading to the deposition of clot. A platelet-rich thrombus is observed in arterial thrombus. In contrast, venous stasis and changes in blood composition (leading to hypercoagulability) can incite the formation of thrombus from local procoagulant events, including small endothelial disruptions at venous confluences, saccules, and valve pockets.