Fig. 17.1
Timeline of medical device development
There are various types of heart valve clinical trials, from trials where a novel valve technology is being used for the first time (“first in human” studies) to post-market trials in which a valve therapy has obtained regulatory approval in the given geography, but is studied further to examine long-term effects and/or to obtain more specific information about the overall therapy. In other words, clinical evidence is vital not only to demonstrate safety and effectiveness of a device therapy in humans but also to further examine how well the device works compared to other devices and/or concomitant treatments. In the specific case of a newly developed heart valve, studies will often be designed to compare the new valve against the native valve, other heart valve devices, and/or standard-of-care treatments. This chapter provides a general overview of the present state of human heart valve clinical trials, including an overview of (1) the current positions of regulatory bodies that oversee trials, (2) the specific features of a trial design, and (3) the many necessary considerations involved in the proper implementation of heart valve clinical trials.
For any designed heart valve trial, the following groups/individuals can be identified, each with their specific role(s) (Fig. 17.2):
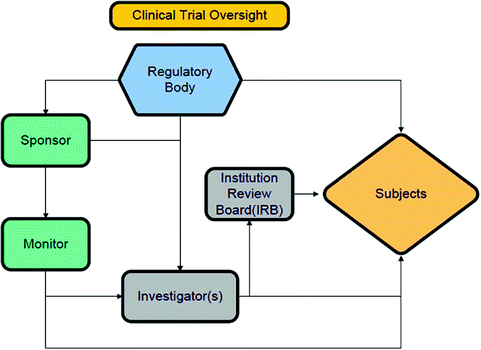
Fig. 17.2
Clinical trial oversight
Sponsor(s):
The developer of the technology seeking approval for market release.
Investigator(s):
Non-biased individuals that will not only implant/deploy the novel technology, but will also be responsible for individual patient follow-ups. In some cases, investigators can also develop with their own field clinical trial(s).
Monitor(s):
Individuals responsible to ensure that the trial is performed in an ethical and proper fashion. They usually work for the sponsor and make frequent visits to participating institutions to review data and regulatory documents. Regulatory bodies also have their own process for auditing sponsors and investigators through their Bioresearch Monitoring group(s).
Institutional Review Board (IRB)/Ethics Committees (ECs):
The overseeing body at a given institution that is ultimately responsible for ensuring that the clinical protocol is appropriate, and that the institutional investigators perform the study in an proper and ethical manner. These boards may have different names according to the institutional structure.
Subjects:
Individual patients who were deemed appropriate to be enrolled (meeting all inclusion and none of the exclusion criteria) into the planned clinical trial and who provided informed consent to participate.
17.2 Regulatory Bodies
Regulations and the regulatory bodies that govern heart valve clinical trials play an important role in how heart valve technologies reach the market. A solid partnership between a sponsor and regulatory body, aided by clear communication, can affect whether technology can reach the market in an expeditious manner. Regulatory bodies are important as they ensure there is consistency in clinical trials and that they are run properly in order to provide the supportive scientific evidence required. Specifically, there are numerous regulatory bodies that provide oversight for heart valve clinical trials throughout the world. A brief overview of regulatory bodies from three different geographies follows, yet our discussion focuses mainly on the Food and Drug Administration (FDA) in the United States.
17.2.1 Food and Drug Administration (United States)
The FDA oversees clinical trials exclusively within the United States. The FDA’s mission statement consists of two primary parts: (1) promoting public health by promptly and efficiently reviewing clinical research and taking appropriate action on marketing of regulated products in a timely manner; and (2) protecting public health by ensuring a reasonable assurance of safety and effectiveness of devices intended for human uses [1]. The Center for Devices and Radiological Health is the branch that oversees medical devices (including heart valves).
In the United States, there are three regulatory classes of devices based on the considered levels of risk involved with a given device. All class I–III devices are subject to general controls, meaning the FDA looks at factors like labeling, registrations, etc. Class I devices have the lowest amount of risk and regulatory controls (e.g., devices such as elastic bandages and surgical gloves). Class II devices must meet specific performance standards in addition to all class I requirements (e.g., devices such as surgical drapes). Most stringently, class III devices require premarket approvals (PMAs) to ensure both their safety and effectiveness. As such, class III devices are also considered as the riskiest category of devices, and include implantable pacemakers and heart valves.
It should be noted that the FDA regulations for medical device products are detailed in Title 21 of the Code of Federal Regulations (CFR). The most applicable parts of CFR 21 that apply to heart valve clinical trials include Part 812 (Investigational Device Exemption, or IDE) and Part 814 (PMA). Most new heart valves are required to undergo IDE clinical trials before receiving FDA approval.
More specifically, according to the FDA’s Heart Valves—Investigational Device Exemption (IDE) and Premarket Approval (PMA) Applications Draft Guidance (dated January 20, 2010): “a replacement heart valve is a device intended to perform the function of any of the heart’s natural valves” [2]. A replacement heart valve is defined as a pre-amendment type device, that is, a device marketed prior to passage of the Medical Device Amendments to the Federal Food, Drug, and Cosmetic Act (the Act).
Furthermore, this FDA’s Draft Guidance explains that clinical trials are needed to evaluate most new replacement heart valve designs and also recommends that clinical investigations of such a replacement heart valve are executed by following the methods described in ISO 5840:2005 or an equivalent document. Specifically, the document ISO 5840:2005 is a guide for cardiovascular implants and valve prostheses provided by the International Organization for Standardization (ISO) [3].
17.2.2 Other Regulatory Bodies
In Europe there are various notified bodies that provide oversight of clinical trials. The most prevalent regulatory oversight applies to the 27 countries in the European Economic Area; these are countries required to obtain a CE mark (“Conformité Européenne” or European Conformity). Importantly, the criteria to receive a CE mark in Europe are notably different than receiving FDA approval. As mentioned previously, to receive approval for a new heart valve technology in the United States, the manufacturer must demonstrate the device to be reasonably safe and effective. To receive approval to release a device to market in the European Union, the manufacturer must demonstrate that the heart valve is safe and that it performs in a manner consistent with the manufacturer’s intended use [4]. Interestingly, given these differences between geographic regulatory approvals, most manufacturers typically receive approval in Europe or other countries before the United States. Moving into other geographies poses different obstacles which may influence the intended quality and importance of every clinical trial completed for the new device.
17.2.3 Good Clinical Practice Oversight
Similar to the importance of following Good Manufacturing Practice (GMP) and Good Laboratory Practice (GLP) when prototyping heart valves, it is important to follow guidelines for how to appropriately conduct clinical studies that could affect the safety and well-being of human participants. Good Clinical Practice (GCP) was developed by a collaborative group of regulatory authorities of various worldwide geographies including the European Union, Japan, and the United States by the International Conference on Harmonisation. GCP was finalized in 1996 and became effective in 1997; it provides international assurance that data and results of clinical investigations are credible and accurate, and that the rights, safety, and confidentiality of participants in the clinical research studies are respected and protected. More specifically, GCP consists of 13 principles (Table 17.1).
Ethics |
·Ethical conduct of clinical trials |
·Benefits justify risks |
·Rights, safety, and well-being of subject prevail |
Protocol and science |
·Nonclinical and clinical information supports the trial |
·Compliance with a scientifically sound, detailed protocol |
Responsibilities |
·Institutional Review Board/Independent Ethics Committee approval prior to initiation |
·Medical care and decisions by qualified physician |
·Each individual qualified (education, training, experience) to perform his/her tasks |
Informed consent |
·Freely given from every subject prior to participation |
Data quality and integrity |
·Accurate reporting, interpretation, and verification |
·Protects confidentiality of records |
Investigational products |
·Conform to good manufacturing practice and used per protocol |
Quality control/quality assurance |
·Systems with procedures to ensure quality of every aspect of the trial |
17.3 The Generalized Clinical Trial Cycle/Process
Addressing all aspects of a clinical trial in depth is an enormous undertaking and beyond the scope of this chapter; thus, the following sections will highlight some of the foundational methods and processes of a typical heart valve clinical trial. As you can see in Table 17.2, there are many tasks that need to be addressed with the development and execution of a clinical trial. It is important to note that some of these tasks may occur simultaneously.
Table 17.2
Standardized clinical research process
1.Prepare a clinical plan |
2.Recruit investigators |
3.Prepare protocol |
4.Prepare case report forms |
5.Prepare informed consent form |
6.Perform investigator site visit |
7.One-on-one investigator reviews, including clinical plan, protocol, case report forms, and informed consent form |
8.Obtain an investigator agreement |
9.Obtain IRB approvals for each participating institution |
10.File an IDE |
11.Obtain an IDE approval |
12.Perform periodic investigator meetings |
13.Conduct the clinical study, i.e., a multicenter study |
14.Monitor the multicenter study |
15.Conclude study |
16.Compile data from each institution |
17.Analyze overall collected data |
18.Write final clinical report |
17.3.1 Features of a Trial Design for a Newly Developed Heart Valve
Prior to planning and execution of a heart valve trial, it is important to research and understand all current published information and relevant heart valve trial data. There is much to be gained from knowing the details of previous trial designs as well as the subsequent outcomes associated with those trials. For gaining FDA approval for any cardiac device, the importance of clinical evidence cannot be stressed enough. In other words, in the beginning stages of planning a clinical trial design, associated publications and previous research can help shape important components for the new trial, such as patient inclusion/exclusion criteria, statistical designs employed in such trials, and/or the general patient populations to be studied.
A well-controlled clinical investigation includes a clear objective and defined methods of analyses. More specifically, the objective should address the proposed medical claims for the investigational device and these should be refined to specifically address the safety and effectiveness of the heart valve in a defined population. Next, it is important to structure a trial so there can be a valid comparison to controls. For example, in current transcatheter valve therapy trials, the new therapy (transcatheter valves) is directly compared to a standard open-heart valve surgery. In other words, a control group in such a trial gives the trial results a meaningful comparison to an existing therapy or treatment. Often, an appropriate control group can be identified by performing a careful and thorough literature search. Furthermore, performing prior research on the specific disease or conditions that the newly designed heart valve will intended to treat is equally important in order to understand the natural progression of the disease or condition and the current benefits or limitation of other treatments. It should be noted that often this step of researching the disease or condition is completed in earlier phases of prototyping of the actual heart valve, but it is recommended that it be reviewed once again just prior planning your clinical trial. Finally, literature searches on similar treatments/heart valves can also assist in identifying the appropriate disease populations and justifying the inclusion and exclusion criteria for the trial.
When the patient population and treatment/control cohorts are clearly identified, one needs to consider the next set of factors to impact the ultimate design. First, the type of trial design must be determined, whether it is randomized, blinded, or double-blinded. Each of the designs may strengthen the significance of the obtained results, while also minimizing bias and providing comparability of groups. Well-defined trial endpoints are also of great significance for the overall success of a clinical trial. For example, typical heart valve trial endpoints should encompass both safety and effectiveness measures. Note that adverse events often comprise the safety endpoint for a given trial. Typically, effectiveness endpoints are found in the form of the presence or absence of a clear, definite effect on a patient, e.g., in a heart valve trial this may include death or the resultant effective orifice area (EOA). Table 17.3 provides a list of key steps to consider in designing a clinical trial for the development of a new heart valve technology.
Table 17.3




General steps in the development of a clinical study design
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
