16

Churg-Strauss Syndrome,
Microscopic Polyangiitis, and Other
Forms of Pulmonary Vasculitis
There is no entirely satisfactory classification of systemic vasculitis, and this is equally true of vasculitis involving the lung. The usual approach to classification combines size of vessel(s) predominantly affected with clinical features and laboratory results, including morphologic findings. Table 16–1 lists the various types of vasculitis that may be seen in the lung, categorized by the size of the vessel involved and the pathologic reaction patterns, and Table 16–2 gives the frequency of these entities. Most forms of vasculitis that involve the lung are categorized as “small-vessel vasculitis,” meaning that, in the systemic circulation, arterioles, capillaries, and venules are primarily affected, although small arteries and veins may be involved as well. In the lung, small-vessel vasculitis commonly involves pulmonary arterial branches that run alongside bronchioles as well as smaller vessels. In North America, lung involvement by medium- and large-vessel vasculitis is distinctly uncommon, although Behçet’s syndrome and Takayasu arteritis are seen with reasonable frequently in other parts of the world (discussed later). Only a handful of cases of giant-cell arteritis involving the lung have been described. Whether macroscopic polyarteritis nodosa ever involves the lung is disputed (discussed later).
Fig. 16–1 shows an algorithm for approaching small-vessel vasculitis in the lung using morphologic, laboratory, and clinical data. This chapter considers primarily Churg-Strauss syndrome and microscopic polyangiitis. Wegener’s granulomatosis is discussed in Chapter 15, and other forms of vasculitis that typically present with pulmonary hemorrhage and that are characterized by capillaritis are covered in Chapter 12. Some of the more uncommon types of pulmonary vasculitis, particularly those involving large and medium-sized vessels, are briefly discussed at the end of this chapter.
The history of vasculitis (reviewed elsewhere1) as a recognized entity dates back to Kussmaul and Maier, who in 1866 described “periarteritis nodosa,” a term later changed to “polyarteritis nodosa” (PAN). PAN typically affected medium-sized arteries and often resulted in the formation of small aneurysms, but for the better part of a century virtually all cases of vasculitis were labeled PAN, even if completely different sizes and types of vessels were involved and aneurysms were not obvious. In the 1930s, 1940s, and early 1950s, investigators began to distinguish several clinically and pathologically different groups of vasculitis. Wegener’s granulomatosis was originally described in the 1930s and further codified by Goodman and Churg in 1954, and Churg-Strauss syndrome was promulgated as an entity separate from PAN in 1951 (discussed later). In the late 1940s, Dawson and colleagues described what they believed was a microscopic form of PAN in which a necrotizing glomerulonephritis was also found. At about the same time Zeek et al described what they referred to as “hypersensitivity angiitis,” an entity that primarily affected small arteries, arterioles, venules, and capillaries, and that in some instances was associated with a necrotizing glomerulonephritis. The condition described by Dawson and colleagues and some of the cases described by Zeek et al would now be classified as microscopic polyangiitis; other cases reported by Zeek et al now would be viewed as cutaneous leukocytoclastic vasculitis.
In 1954 Goodman and Churg reviewed the literature and their own experience and suggested that Churg-Strauss syndrome, Wegener’s granulomatosis, and what is currently referred to as microscopic polyangiitis were related forms of vasculitis distinct from polyarteritis nodosa. These three diseases are in many respects both clinically and morphologically similar (see Differential Diagnosis, later in this chapter) and their relationship has been strengthened by the observation that all three usually do not have deposition of immunoglobulins in tissue (and hence are labeled “pauci-immune”) and frequently are associated with antineutrophil cytoplasmic antibodies (ANCAs). They affect predominantly small arteries, arterioles, capillaries, venules, and veins, and therefore fall under the general heading of small-vessel vasculitis.
Disease | Pathologic Reaction Patterns in Lung |
|---|---|
Large-vessel vasculitis (aorta and large branches of the pulmonary artery) | |
Giant-cell arteritis | Giant-cell or granulomatous arteritis |
Takayasu arteritis | Giant-cell or granulomatous arteritis with aneurysm formation and/or vascular obliteration, infarction, pulmonary hypertension |
Behçet disease | Arteritis with aneurysm formation and thrombotic vascular obliteration, infarction, pulmonary hypertension |
Medium-sized vessel arteritis | |
Polyarteritis nodosa | Involvement of lung disputed, but putative microscopic picture is that of a necrotizing arteritis in bronchial or pulmonary arteries |
Small-vessel vasculitis: immune complex mediated | |
Vasculitis in lupus erythematosus | Capillaritis with hemorrhage Occasionally involvement of larger vessels |
Vasculitis in rheumatoid arthritis | Necrotizing small vessel vasculitis Occasionally capillaritis with hemorrhage |
Vasculitis associated with cryoglobulinemia | Capillaritis with hemorrhage Occasionally involvement of larger vessels |
Henoch-Schönlein purpura | Capillaritis with hemorrhage |
Goodpasture’s syndrome (anti-GBM disease) | Capillaritis with hemorrhage/interstitial fibrosis in long-standing cases |
Small-vessel vasculitis: pauci-immune (usually ANCA positive) | |
Wegener’s granulomatosis | Necrotizing vasculitis involving small arteries, arterioles, venules, veins, and capillaritis with hemorrhage; often granulomatous |
Churg-Strauss syndrome | Necrotizing vasculitis involving small arteries, arterioles, venules, veins, and capillaritis with hemorrhage; infiltrate is eosinophilic, often granulomatous |
Microscopic polyangiitis | Capillaritis with hemorrhage and sometimes involvement of larger vessels, not granulomatous |
Drug-induced vasculitis | Capillaritis with hemorrhage |
Miscellaneous forms of small-vessel vasculitis and mimics of vasculitis | |
Vasculitis associated with TB and fungal infections | Granulomatous vasculitis associated with necrotizing granulomas |
Vasculitis of pulmonary hypertension | Necrotizing vasculitis of small arteries with other changes of pulmonary hypertension |
Antiphospholipid antibody syndrome | Vascular thrombosis without significant inflammation of vessel wall; pulmonary hemorrhage with or without capillaritis |
ANCA = antineutrophil cytoplasmic antibody; GBM, glomerular basement membrane; TB = tuberculosis.
More recently it has been recognized that other forms of small-vessel vasculitis, forms that are morphologically similar to the ANCA-positive vasculitides, are ANCA negative and instead characterized by the presence of both circulating immune complexes and immune complexes deposited in the kidneys and lungs as well as other organs. In the lung the most common of these immune-complex, small-vessel vasculitides are capillaritis associated with lupus and Goodpasture’s syndrome (antiglomerular basement membrane disease), but a variety of other conditions produce a similar picture (Tables 16–1 and 16–2; Fig. 16–1) (see Chapter 12).
Lung Involvement Frequent | Lung Involvement Uncommon |
|---|---|
Wegener’s granulomatosis (90% of cases)a | Cryoglobulinemiac |
Churg-Strauss syndrome (70% of cases)a | Henoch-Schönlein purpurac |
Microscopic polyangiitis (50% of cases)a | Drug vasculitisc |
Pulmonary renal syndromesb | Takayasu arteritisd |
Wegener granulomatosis | Behçet diseased |
Microscopic polyangiitis | Giant-cell arteritisd |
Collagen vascular diseases | Macroscopic polyarteritisd |
Goodpasture’s syndrome |
|
aPercentage of cases that involve the lung at some time in the course of the disease.
bCombination of glomerulonephritis and pulmonary hemorrhage.
cMay manifest as pulmonary renal syndrome.
dLarge- and medium-vessel vasculitis.
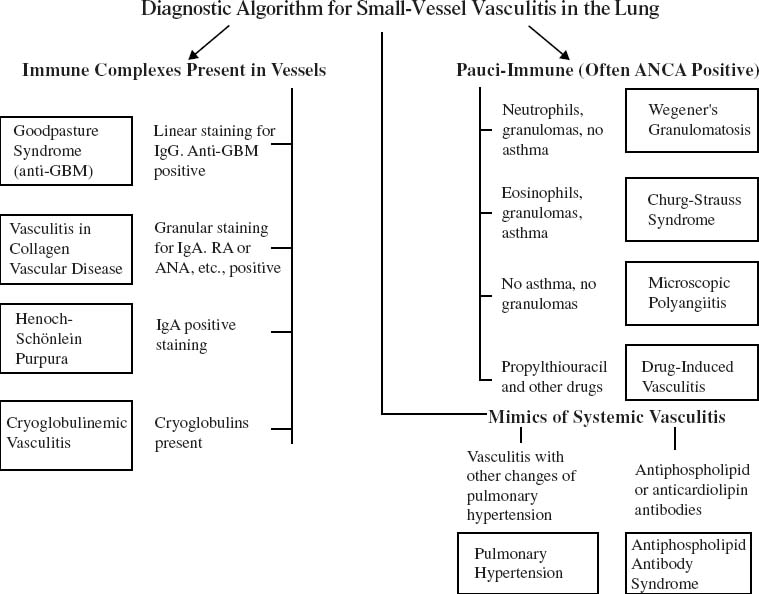
FIGURE 16–1 Diagnostic approach to small-vessel vasculitis in the lung.
Churg-Strauss Syndrome
History and Definition
Churg-Strauss syndrome (CSS) comprises asthma, eosinophilia, and, in its full-blown form, eosinophil-rich systemic vasculitis. However, as will become apparent, many cases of CSS, especially the fairly early ones readily amenable to treatment, do not have overt vasculitis, and a much broader definition is required to include these cases.
Patients who would probably fulfill the current criteria for CSS were described by Rackeman and Greene2 in 1939 as having polyarteritis nodosa, asthma, and eosinophilia. Additional similar patients were described by Harkavy in 1941 and 1943.3,4 The first formal description of CSS was made by Churg and Strauss5 in 1951, based on 13 autopsied patients, under the title “Allergic Granulomatosis, Allergic Angiitis, and Periarteritis Nodosa.” As emphasized by Churg and Strauss, the findings common to all patients were (1) history of asthma, (2) serum and tissue eosinophilia, (3) necrotizing vasculitis, and (d) a granulomatous response to eosinophilic necrosis.
Despite the apparent homogeneity of findings in the original patients, further experience has shown that the exact definition of CSS is a problem, in part because CSS, like most forms of vasculitis, is very protean in its manifestations, and in part because, as noted, some early cases do not demonstrate vasculitis. As well, the original cases predated the use of exogenous steroids, but most asthmatics now are routinely treated with steroids for their asthma, and in some cases treatment itself changes both the clinical and pathologic appearances of CSS (see Formes Frustes of Churg-Strauss Syndrome, later in chapter).
Although initial studies emphasized pathologic diagnosis, the recognition that CSS could be treated with steroids has led to greater emphasis on clinical diagnosis, with or without biopsy.6,7 Table 16–3 lists the original criteria of Churg and Strauss and three more recent definitions. The American College of Rheumatology (ACR) definition8 incorporates allergic rhinitis as well as asthma, because it has become apparent that a very large proportion of CSS patients have allergic rhinitis and that this in fact is often the earliest manifestation of disease.6,7 The Lanham et al6 definition emphasizes the presence of overt vasculitis, whereas the ACR definition incorporates both vasculitic features such as neuropathy and nonvasculitic features such as pulmonary infiltrates or extravascular eosinophil infiltration of tissues on biopsy. The ACR criteria are claimed to produce a specificity of >99% and sensitivity of ~85% if four of six criteria are met, although the ACR criteria were actually developed to use on patients with a definite clinical diagnosis of vasculitis, and give lower sensitivities when the diagnosis of vasculitis is not clear. These definitions all predate the widespread use of ANCA testing, but a positive ANCA result provides strong support of the diagnosis of vasculitis (see below). Some familiarity with these definitions is of use to the pathologist asked to indicate whether a biopsy shows findings compatible with CSS.
Churg and Strauss5 | History of asthma |
| Blood and tissue eosinophilia |
| Necrotizing vasculitis |
| Necrotizing granulomas centered on necrotic eosinophils |
Chapel Hill Consensus Conference9 | Asthma |
| Eosinophilia |
| Necrotizing vasculitis |
| Granulomatous inflammation associated with tissue eosinophilia |
Lanham et al6 | Asthma |
| Eosinophilia >1.5 × 109/L |
| Vasculitis involving at least two organs |
American College of Rheumatology8 | Asthma |
| Paranasal sinus abnormalities |
| Eosinophilia >10% |
| Neuropathy (mono- or poly-) |
| Pulmonary infiltrates |
| Biopsy containing a blood vessel with extravascular eosinophils |
Incidence of Churg-Strauss Syndrome
Attempts to determine the incidence of CSS are also fraught with difficulty, in part because varying definitions produce varying incidence rates and because the disease is uncommon. Kurland et al10 estimated an incidence of 4.0/million in Olmsted County, Minnesota, based on the observation of only one case. Watts et al,11 using the adult population of the Norwich Health Authority, found an incidence of 2.4 cases/million persons/year (Table 16–4). This was considerably less than the incidence of Wegener’s granulomatosis or systemic rheumatoid vasculitis, but comparable to the incidence of microscopic polyangiitis. Of particular interest is the report by Martin et al.12 Using prescription-related event monitoring, they found that for cohorts of antiasthma-drug users, the prevalence rate of CSS was 64.4 cases/million patient-years of observation, compared with cohorts monitored for other types of prescriptions where the prevalence was 1.8 cases/million patient-years of observation. This finding serves to emphasize the fact that, although CSS is a fairly uncommon disease in the general population, it is relatively frequent in asthmatics.
Antineutrophil Cytoplasmic Antibodies and the Pathogenesis of Churg-Srauss Syndrome
Antineutrophil cytoplasmic antibodies (ANCAs) were first recognized to be associated with vasculitis in the early 1980s. The original tests were done by applying a patient’s serum to smears of normal alcohol-fixed human neutrophils and then using a second fluorescent-labeled anti-human immunoglobulin to detect bound antibody. Using this approach, the two basic patterns of reaction are cytoplasmic (c-ANCA), characterized by cytoplasmic fluorescence with greater intensity between the lobes of the neutrophil nucleus, and perinuclear (p-ANCA), characterized by bright fluorescence along the nuclear membrane.14–16 The immunofluorescent tests are often difficult to interpret, and considerably greater specificity has been achieved using enzyme-linked immunosorbent assay (ELISA) directed against the neutrophil enzymes myeloperoxide, which corresponds to fluorescent p-ANCA, or proteinase 3, which corresponds to fluorescent c-ANCA, these tests, albeit still not entirely standardized,14 are the procedures of choice. Using this approach, there is a reasonably high correlation between positive ANCA titers and the presence of some forms of small-vessel vasculitis, namely Wegener’s granulomatosis, which is c-ANCA positive in up to 90% of active cases, and microscopic polyangiitis and CSS, both of which are reported to show p-ANCA positivity in up to ~70% of cases (Table 16–5). Some types of drug-induced vasculitis are also ANCA positive (see Drug-Induced Vasculitis, later in this chapter). A proportion of cases of pauci-immune rapidly progressive glomerulonephritis (RPGN) are ANCA positive, although these probably represent renal-limited microscopic polyangiitis. The exact specificity and sensitivity of ANCA tests are the subject of some controversy, and the interested reader is referred elsewhere for detailed discussion;14–18 these authors also consider the equally controversial question of whether ANCA can be used to follow disease activity.
Watts et al 199511 | Systemic rheumatoid vasculitis | 12.5 cases/million adults/year |
Norwich Health Authority10 | Wegener’s granulomatosis | 8.5 cases/million adults/year |
| Churg-Strauss syndrome | 2.4 cases/million adults/year |
| Microscopic polyangiitis | 2.4 cases/million adults/year |
Kurland et al 1984 Olmsted County, MN10 | Churg-Strauss syndrome | 4.0 cases/million/year |
Martin et al 1999 Prescription event monitoring12 | Churg-Strauss syndrome |
|
Not known to be asthmatic | 1.8 cases/million/year | |
| Known asthmatics | 64.4 cases/million/year |
Loughlin et al 200213 | Churg-Strauss syndrome |
|
| Asthmatics who did not use leukotriene receptor antagonists | 0 to 67 cases/million/year depending on criteria |
c-ANCA (anti–proteinase 3) | Usually found in Wegener’s granulomatosis, occasionally in CSS and microscopic polyangiitis; rare in normals |
p-ANCA (antimyeloperoxidase) | Commonly in CSS and microscopic polyangiitis, occasionally in Wegener’s granulomatosis; some cases of pauci-immune rapidly progressive glomerulonephritis (RPGN) |
| Occasionally in poststreptococcal GN, HIV infections, myelodysplasia |
| Some cases of antiglomerular basement membrane disease (Goodpasture’s syndrome) |
| Drug-induced vasculitis (propylthiouracil, hydralazine) |
| Up to 5% of normal blood bank samples |
Atypical ANCA (sometimes antilactoferrin or cathepsin G or β-glucuronidase) | Chronic inflammatory bowel disease |
| Primary biliary cirrhosis |
| Sclerosing cholangitis |
| Amoebiasis |
What these discussions do make clear is that ANCA staining or reactivity is really of use only in a clinical setting where there is a reasonable probability that a patient has vasculitis. As a screening test, ANCA is misleading: c-ANCA and especially p-ANCA are found in normal blood samples from a small fraction of the otherwise apparently healthy general population.14 Both fluorescent p- and c-ANCA patterns and so-called atypical ANCAs, which show unusual staining patterns and have antigenic specificities other than proteinase 3 or myeloperoxidase, are found in a variety of nonvasculitic diseases (Table 16–5). But in the setting of a patient with features of CSS, the finding of a positive antimyeloperoxidase or anti–proteinase 3 ANCA is strongly supportive of the diagnosis.
The pathogenesis of CSS is poorly understood, but recent studies on the pathogenesis of vasculitis in general have focused on the role of ANCA.15,19 The question of whether ANCAs by themselves are pathogenic is controversial, but new experimental evidence20 indicates that administration of ANCAs to mice can produce glomerulonephritis. Moreover, if polymorphonuclear neutrophils (PMNs) are primed, for example with low doses of tumor necrosis factor-α (TNF-α), a process that exposes proteinase 3 and myeloperoxidase on the cell surface, and then treated with ANCA, the cells release reactive oxygen species and proteases. This process also leads to upregulation of the production of leukotriene B4 and interleukin-8 (IL-8), both neutrophil chemoattractants. As well, primed neutrophils can detach and destroy endothelial cells in the presence of ANCA, and at least some types of endothelial cells express ANCA target antigens on their surface. In an inflammatory milieu endothelial cells also upregulate adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), leading to further neutrophil adhesion. Thus, ANCA can cause release of tissue-damaging agents (oxidants, proteases) and the recruitment of PMN, and probably also lead to vascular injury that appears, pathologically, as vasculitis. In addition, there is evidence that patients with vasculitis may develop antiendothelial antibodies that presumably serve to increase endothelial damage.19
Although there are good experimental data to support a major role for ANCA in producing vasculitis, the mechanisms behind the appearance of ANCA are unknown. Experimentally, some drugs (e.g., propylthiouracil) appear to act as haptens and can stimulate formation of ANCA. It has been suggested that low-grade infections may lead to the formation of antibodies with proteinase 3 or myeloperoxidase specificity.19 Why eosinophils predominate in CSS and PMN in the other ANCA-associated forms of vasculitis is unclear. Similarly, the process that leads to PMN priming (assuming the models are correct) is unknown, but speculation frequently centers on low-grade infections, and this idea is supported by the observation that co-trimoxazole suppresses disease in some cases of Wegener’s granulomatosis.14–19
In most patients who develop CSS, there is no obvious precipitating event, although typically these patients have moderate to severe asthma. The de novo development of CSS has been reported in asthmatic patients undergoing desensitization treatments or receiving vaccinations against diverse infectious agents including tetanus, influenza, and tuberculosis;21,22 as a drug reaction;21,23 and in parasitic infections.24,25 CSS may also appear for the first time in asthmatics maintained on steroids when their steroids are tapered or discontinued (see Formes Frustes, later in this chapter).
Natural History: The Early (Prodromal, Prevasculitic), Vasculitic, and Postvasculitic Phases of Churg-Strauss Syndrome
The original patients described by Churg and Strauss5 all had florid vasculitis, but it has become apparent that many CSS patients experience an early (also called prodromal6,7 or prevasculitic) phase before the development of vasculitis (Table 16–6). Recognizing patients with early disease is important because they appear to respond particularly well to steroids and to have an excellent prognosis (discussed later).
Lanham et al6 summarized 138 cases in the literature and 16 of their own, and suggested that the typical sequence of events is, initially, the appearance of clinically overt allergic rhinitis, followed by the development of asthma several years later, and finally the development of vasculitis several years after that. In their literature summary the mean age at which rhinitis occurred was 28, for asthma 35, and for vasculitis 38. Some authors also describe what they refer to as a postvasculitic phase in successfully treated patients:6,7 asthma and allergic rhinitis are still present, and there is no active vasculitis, but persisting neuropathy and hypertension may be seen.
Clinical Features in the Early Phase
Allergic rhinitis was (in retrospect) the earliest manifestation of CSS in ~70% of the cases summarized by Lanham et al6 and 60% of the cases of Guillevin et al.21 The disease is often severe and may require repeated nasal polypectomies to relieve obstruction and sinusitis; radiographic surveys suggest that the true incidence of sinusitis is even higher.6 Asthma typically first develops in adulthood rather than childhood, usually after the onset of allergic rhinitis. The reported interval between the appearance of asthma and overt CSS varies from months to over 30 years, and in rare cases asthma first appears after the onset of vasculitis.21 Many patients have a history of increasingly difficult-to-control asthma, and this may be part of early-stage disease,26 although by itself neither asthma nor allergic rhinitis is specific for CSS. Oddly enough, the asthma sometimes becomes milder once overt CSS develops.6
Allergic rhinitis and/or asthma |
Blood eosinophilia |
Extravascular tissue infiltration by eosinophils (may be diagnosed by fine-needle aspirate) |
High levels of lavage eosinophilia |
Chronic eosinophilic pneumonia |
Eosinophilic gastroenteritis |
Enlargement of lymph nodes, salivary glands, or other organs caused by eosinophilic infiltration |
No vasculitis clinically or on biopsy |
Blood eosinophilia may be present in the early, vasculitic, or postvasculitic phase of CSS, and is found at some time in all patients with CSS. Eosinophilia tends to wax and wane and does not correlate clearly with other manifestations of disease activity; serum immunoglobulin E (IgE) levels are claimed to show better correlations.26 Lanham et al6 suggested that eosinophilia >1.5 109/L (at some point) is a required feature for diagnosis. This value is much higher than the blood eosinophilia sometimes found in asthmatics who do not have CSS, and also considerably higher than is seen in the rare cases of Wegener’s granulomatosis with associated eosinophilia.27 In fact, the eosinophil count in CSS cases is often considerably greater than the cutoff suggested by Lanham: in the original cases of Churg and Strauss5 levels usually exceeded 5 × 109/L. Blood eosinophil levels may revert to normal or near-normal values, but tissue infiltration by eosinophils can still be present in this situation.7 It should also be remembered that steroid therapy for asthma can suppress blood eosinophilia.
Fever and malaise may be present in the early stage of CSS,26 but fever and weight loss are also extremely common presenting signs in the vasculitic phase.21
Extravascular tissue infiltration by eosinophils is the characteristic feature of the early phase of CSS and may occur in any organ. Chest radiographs often reveal the fleeting airspace infiltrates referred to as Loeffler’s syndrome; this typically is not associated with systemic signs and rapidly disappears.28 However, Loeffler’s syndrome is common in asthmatics and is not specific to CSS. In other early-phase cases there is a radiographic picture of chronic eosinophilic pneumonia with persistent peripheral dense airspace infiltrates (Fig. 16–2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


