Physiopathology of sickle cell anemia. (Source: Adapted from Rees DC et al. Lancet. 2010;376:218–222)
Vaso-occlusive crises are caused by erythrocyte and leukocyte entrapment in the microcirculation, originating vascular obstruction and tissue ischemia. Although this process requires HbS polymerization, the trigger event for vascular obstruction is a type of inflammation. This inflammation results from an interaction between the erythrocyte and vascular endothelium, causing obstruction and ischemia episodes, which are followed by a restitution of the vascular flow, causing tissue damage mediated by reperfusion. Then, oxidative stress is triggered, which causes adhesion molecule overexpression, increasing inflammatory cytokines synthesis and leukocytosis. Hemolysis also contributes to vaso-occlusion. Hemoglobin liberation in plasma, caused by intravascular hemolysis, generates superoxide radicals and hydroxyl, which are potent inhibitors of nitric oxide (NO). This compound is produced under normal conditions in the endothelium and regulates the basal vasodilator tone, inhibits platelets, hemostatic activation, and the expression of adhesion molecules dependent on the nuclear factor kβ (FNkβ). Hb release into the plasma also causes endothelial dysfunction and NO resistance. Hemolysis also liberates arginase-1 in the erythrocyte, which metabolizes arginine into ornithine, exhausting the substrate required to synthesize NO. All of this helps to maintain hypercoagulability, with an increase in the platelet activation and the levels of procoagulant factors in the blood.
It is important to note that acute and chronic inflammatory events happen in the lung because erythrocytes are exposed to relatively low O2 tensions, as well as the slow flow of the cells. The airway and vascular system are in close connection, which eases the transference of inflammatory mediators among each other.
Clinical Manifestation of Sickle Cell Disease
SCD has considerable phenotypical heterogenicity, influenced by genetic and environmental factors. Hb of fetal concentration (HbF), coexistence of other hemoglobinopathies, and certain types of polymorphism in simple nucleotides modulate the risk of certain complications. Among environmental factors, environmental humidity, cold, and pollution negatively influence the patient, and particularly by increasing vaso-occlusive events.
Complications worsen with age. In infants, dactylitis (painful inflammation of the fingers and toes), anemia, hyperbilirubinemia, splenomegaly, and infections in the respiratory tract are common. Among other complications, children may present growth and puberty delay, cognitive alterations, and cerebrovascular accidents. Adults tend to have articular pain, chronic ulcers in the legs, kidney failure, and neurocognitive disorders.
Respiratory problems associated with sickle cell anemia
Pulmonary manifestation | Respiratory symptoms | Causes |
|---|---|---|
Acute chest syndrome | Hypoxemia and dyspnea Crackles Sound reduction in lung fields | Multifactorial |
Asthma | Wheezing Dyspnea | Airway hyperreactivity |
Alterations in lung function | Asymptomatic Hypoxemia | Restrictive and obstructive lung disease |
Obstructive sleep apnea | Flow oximetry reduction during sleep Apnea | Increase of lymph tissue in Amygdale and adenoids |
Day hypoxemia | Hypoxemia Dyspnea | Hemoglobin desaturation Pulmonary fibrosis |
Pulmonary hypertension | Hypoxemia Dyspnea Exercise intolerance | Hemolysis Endothelial dysfunction |
Respiratory Clinical Manifestations, Diagnostic Approach and Treatment
Acute Chest Syndrome
Acute chest syndrome (ACS) is a symptom of sudden pulmonary damage, defined as an infiltration of new consolidated alveoli in chest X-rays, with no evidence of atelectasis, and which involves at least a whole lung segment. Generally, the patient presents with chest pain, fever, tachypnea, wheezing, cough, and hypoxemia. The Cooperative Study of Sickle Cell Disease (CSSCD) reported an incidence of 29% (12.8 episodes for 100 patient-years) in patients with sickle cell anemia type SS. Almost half the patients with sickle cell anemia will present with one episode of acute chest syndrome, which is the second cause of hospitalization, after vaso-occlusive crisis (VOC). This may be the initial presentation, although it can also appear after the first 3 days, in 10% to 20% of the cases during their hospital stay. Children between 2 and 4 years of age have the greatest incidence (25.3 years per patient).
Risk factors for this complication involving having HbSS or HbS/β0, thalassemia, asthma, chronic hypoxemia, low HbF, tobacco smoke exposure, general anesthesia, and surgery, mainly abdominal, and during the winter season. There are multiple causes for ACS. The National Acute Chest Syndrome Study Group (NACSSG) studied the causes in 671 episodes presented in 538 patients. Infections were the main cause in 29% of the cases. It is thought that respiratory infections promote an inflammatory response in the lung. Pneumonia caused by Chlamydia was the most common cause, followed by the pneumonia caused by Mycoplasma, viral pneumonia, and bacterial infections last.
Another cause for the acute chest syndrome is fat embolism. During a bone ischemic event, a piece of the marrow or bone that is detached because of a bone marrow stroke migrates through the blood flow to the lungs. As a consequence, sudden inflammation appears. This syndrome caused by fat embolism tends to have a severe clinical presentation, and it tends to appear with pain, neurological symptoms, thrombocytopenia, and increase in transaminases.
Activation of the secretory phospholipase A2 hydrolyzes the fat emboly in sn-2 position, creating free fat acids and lysophospholipids, which cause lung damage. When arachidonic acid is caused by this hydrolysis, a series of inflammation mediators appear, such as thromboxane, leukotrienes, and prostaglandins. Secretory phospholipase A2 increases (336 ± 209 ng/ml; basal level 10 ± 8.4 ng/ml) during an acute chest syndrome presentation, and this increase can predict the disease within the following 24–48 h. Nevertheless, this analysis is still not commercially available, and it has to be validated as a diagnostic test.
A third mechanism of the ACS is pulmonary strokes. This phenomenon has not been carefully studied, and it could be an exclusion diagnosis. Recently, it has been observed that patients may have pulmonary artery thrombosis. This etiology was discovered in 17% of the cases using multidetector computed tomography (CT). Most of the positive findings (81%) in the study by Mekontso Dessap et al. were partial defects in blood vessels. Patients received anticoagulation treatment. Risk factors involved were thrombocytosis and less evidence of hemolysis.
The clinical presentation of the ACS fluctuates in severity. The physician must be alert to detect the beginning of this process. The typical clinical presentation is characterized by chest or limb pain, and fever, followed by hypoxemia and dyspnea. Sometimes the patient has yellow sputum, probably related to fat embolism. Lung examination may reveal crackles, wheezing, and reduction of lung sounds. It is common to find a reduction of hemoglobin levels of 0.7 g/dl on average, and sometimes thrombocytopenia is also present. Thrombocytopenia is an independent risk factor to suffer from multilobe ACS, as well as for needing assisted ventilation. Although most patients can be treated without problems, 13% may require assisted ventilation. CSSCD investigators reported a mortality of 1.1% of children and 4.3% in adults. Other studies have reported a rate of almost 9% in adults.
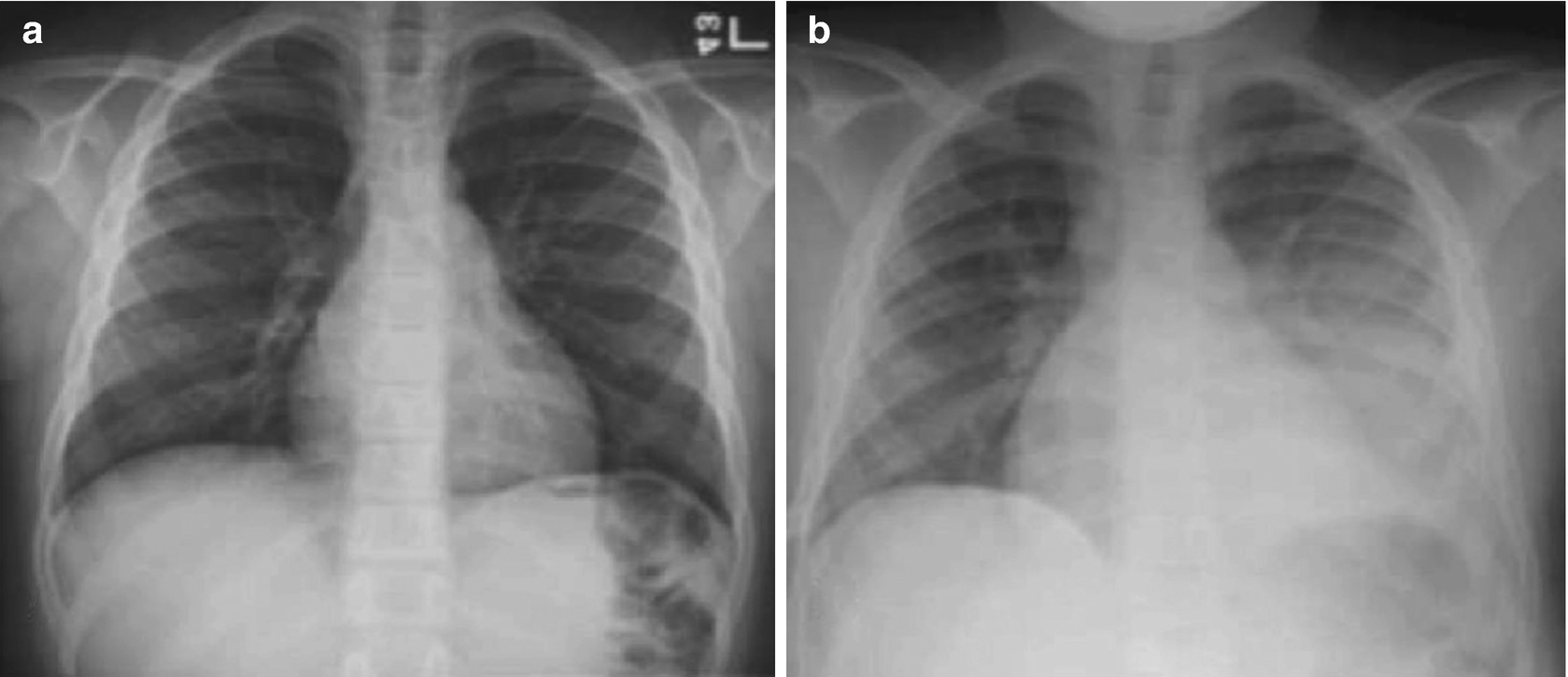
Acute chest syndrome (ACS) shown in chest X-ray in 12-year-old schoolchild with ACS (a) at admittance caused by coronavirus (CoV), and (b) after ACS has been established
It is recommended to repeat the hemogram to diagnose anemia and thrombocytopenia, as well as obtaining a crossed histocompatibility test, in case a transfusion is needed. Obtaining a gasometric analysis will be of use during the treatment, particularly if the patient presents with a case of respiratory distress. If fever is present, a blood culture is required, to identify the bacterial agent in a reduced number of patients (3.5%). The following are not routine tests: sputum culture, D-dimer exam, and computerized tomography.
Bronchoscopy is not necessary for diagnosis and it is not recommended. Nevertheless, in special cases, if the patient’s condition worsens considerably in spite of the treatment, it could help to find a cause such as plastic bronchitis. This complication consists of a mucus blockage caused by bronchial secretions that worsens the hypoxemic state and lung condensations. In this case, the procedure improves the patient’s condition, both clinically and radiographically. Secretion blockages can be removed with saline washing and continuous suction of the blockage, which is then attached to the tip of the bronchoscope; after this, the bronchoscope is removed, with the blockage of the airway attached to it.
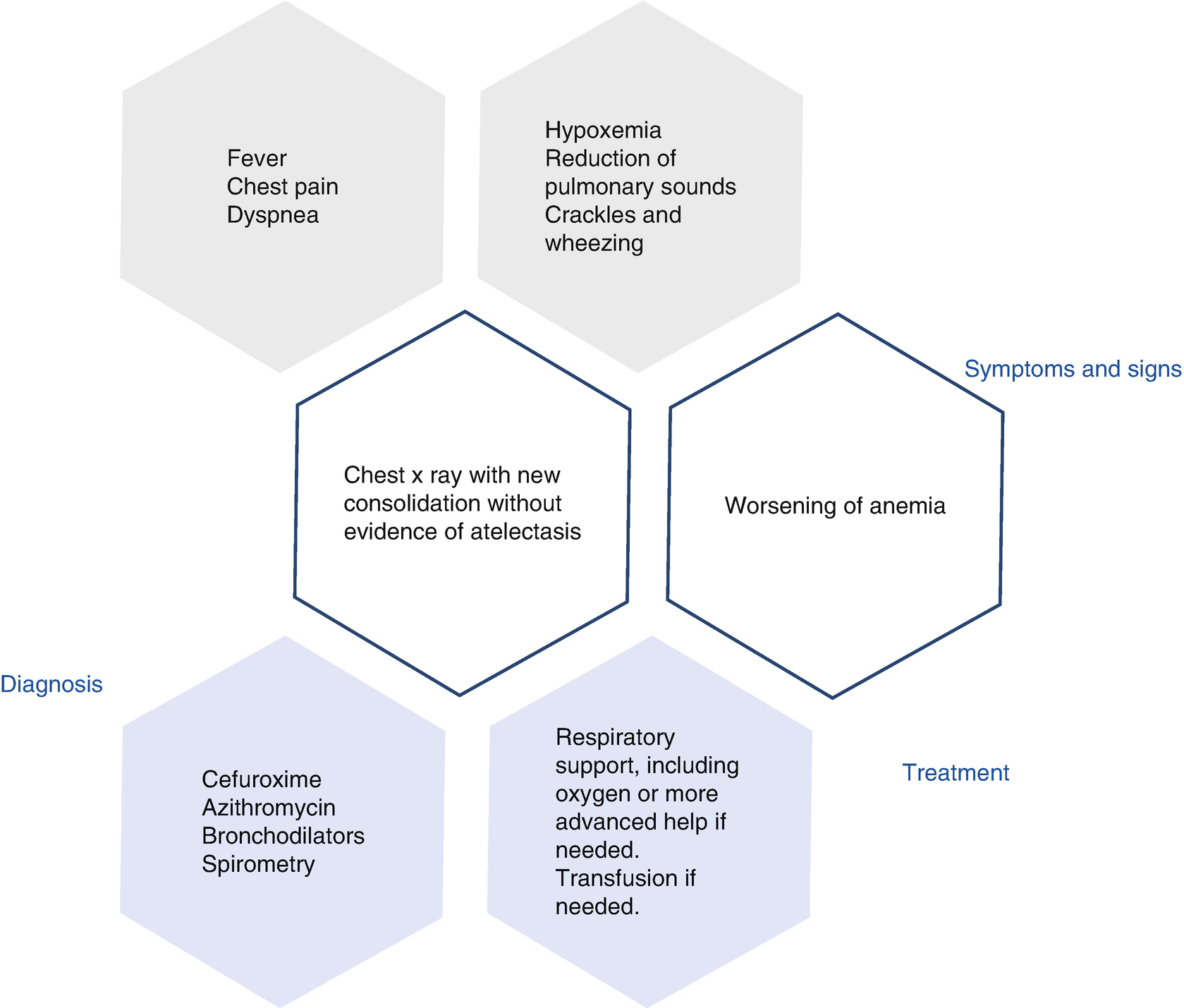
Algorithm of diagnosis and management of ACS
A combination of an IV cephalosporine, such as cefuroxime, with a macrolide such as oral azithromycin, is the standard care. As an alternate regime, in case of allergies, a quinolone or a macrolide with a beta-lactam antibiotic is recommended. Oxygen must be supplied to keep the saturation over 95%. Blood transfusion must not surpass a maximum of Hb 10–11 g/dl to avoid increasing viscosity. This precaution may prevent ACS worsening, and is are recommended for severe anemia or persistent hypoxemia. Transfused red blood cells must be S negative. Blood replacement is recommended for the cases when the initial hemoglobin is high (Hb ≥ 10 g/dl) and the patient requires transfusion. Other indications for blood replacement are severe hypoxemia in spite of oxygen therapy, patients who need an artificial respirator, multilobe ACS, and also if there is no improvement after a simple transfusion.
An echocardiography is recommended for patients who need intensive care to detect lung hypertension. If this is the case, patients must receive intensive treatment with blood replacement.
Because of airway hyperreactivity in SCD, some investigators propose the routine use of bronchodilators, although others only use it for treating wheezing or prolonged expiration.
Use of steroids such as IV dexamethasone 0.3 mg/kg every 12 h at four doses may shorten hospitalization time, but it is associated to relapses in up to 25% of the patients once the treatment is interrupted, and therefore its use is controversial. Although NO seems to be related to the pathology of the hemolysis, as well as improving ACS in preliminary trials, it did not reduce the duration of the vaso-occlusive crisis. Therefore, it had no impact on the development of this syndrome in clinical trials done afterward.
ACS morbidity is wide, both in the short and long term. This syndrome may be conditioned to the development of chronic lung disease, acute lung hypertension during the episode (specially in adults), and respiratory failure (13% of NACSSG patients). Wide lobal affectation, platelets <200,000/mm3, and a history of heart disease are independent prognosis factors for respiratory failure. For difficult cases of respiratory failure that do not respond to conventional treatment, high-frequency oscillator and extracorporeal membrane oxygenation (ECMO) have been used.
Neurological events are other complications during ACS, appearing in 11% of the patients. The most common events are alterations of the mental state, convulsions, and neuromuscular abnormalities. A platelet count under 200,000 mm3 is a risk factor for neurological complications.
The prevalence of acute kidney damage increases with the severity of the chest syndrome. Patients with this syndrome may develop multiple organ failure. Hydroxyurea , a drug that has the capacity to increase HbF, or a chronic regime of monthly transfusions could reduce the recurrence or severity of the episodes. Patients who continue to experiencing episodes even when treated with hydroxyurea or transfusions are candidates for bone marrow transplant, especially if they have a histocompatible sibling.
Pulmonary Function Tests
Spirometry, lung volume, diffusing capacity, or t transfer factor of the lung for carbon monoxide (DLCO), corrected for hemoglobin, and reactivity measures of the reversible airway with bronchodilator are routine lung function tests for patients with SCD. The methacholine parasympathetic agonist challenge test is not done routinely, because of the low risk to induce vasoconstriction and vaso-occlusion.
Lung function
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


