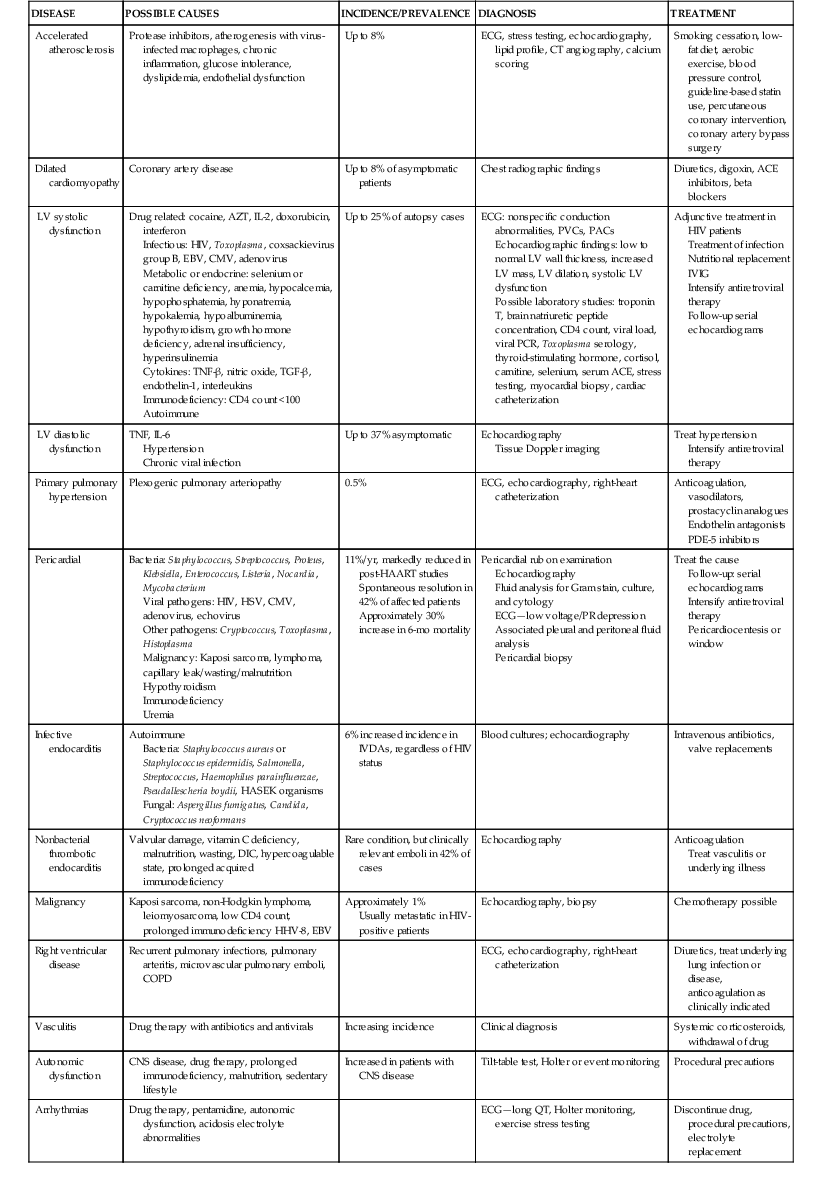
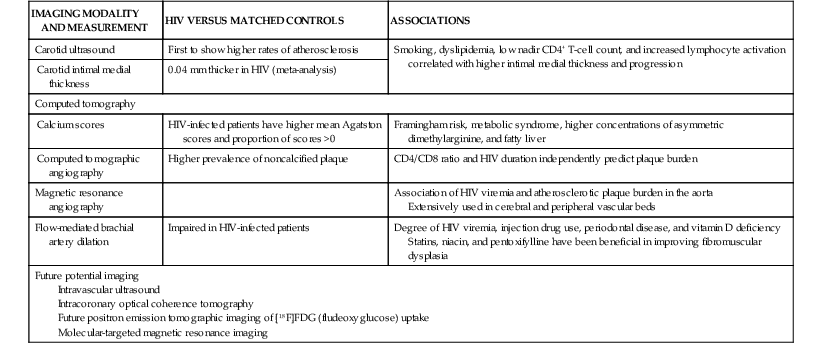
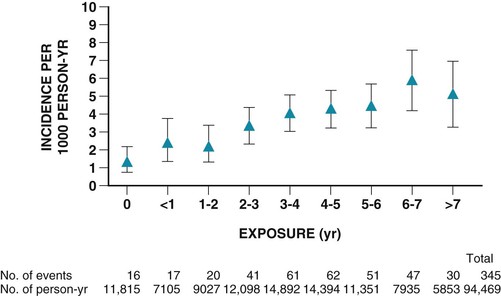
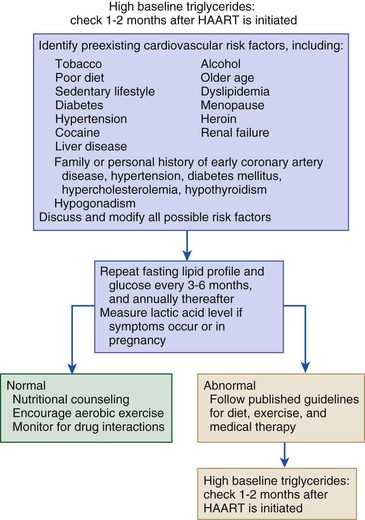
Stacy D. Fisher, Steven E. Lipshultz Infection with human immunodeficiency virus (HIV) is a leading cause of acquired heart disease worldwide and specifically of accelerated atherosclerosis, symptomatic heart failure, and pulmonary arterial hypertension (PAH).1–5 Cardiac complications of HIV infection tend to occur late in the disease in those with acquired immunodeficiency syndrome (AIDS) or prolonged viral infection and are therefore becoming more prevalent as longevity improves.1–5 Multiagent therapies for HIV infection have prolonged life but may also increase later cardiovascular risk and accelerate atherosclerotic disease and events.1,6 Globally, between 31 and 36 million people were living with HIV at the end of 2011,7 including an estimated 0.8% of all those aged 15 to 49 years. Sub-Saharan Africa remains the area most severely affected by HIV infection, with almost 1 in every 20 adults living with HIV and accounting for 69% of all HIV cases worldwide.7 Cardiac abnormalities associated with HIV infection include premature myocardial infarction (MI) or stroke, pericardial effusion, lymphocytic interstitial myocarditis, dilated cardiomyopathy (frequently with myocarditis), left ventricular (LV) diastolic dysfunction, infective endocarditis, and malignancy (myocardial Kaposi sarcoma and B-cell immunoblastic lymphoma) (Table 70-1 and Video 70-1 VIDEO 70-1 Cardiovascular abnormalities in HIV-infected individuals Accelerated atherosclerosis can occur in HIV-infected young adults and children without traditional coronary risk factors (see Chapter 41).8,9 Pronounced coronary lesions were discovered at autopsy in HIV-positive patients 23 to 32 years of age who died unexpectedly, thus prompting research in this area.5,9 Autopsy findings indicated altered histologic characteristics and atherosclerotic plaque with features common to both coronary atherosclerosis and transplant-related vasculopathy. When compared with the general population, affected patients are often younger and more commonly have single-vessel disease in which plaque rupture is the cause of MI.9,10 Acute MI is frequently the first manifestation of atherosclerotic disease (see Chapter 51).11 Inflammation may cause such premature cardiovascular events (Table 70-2). In September 2012, the National Heart, Lung, and Blood Institute (NHLBI) AIDS working group stated that recommendations and research into HIV-related effects on the heart should be priorities and noted the complex interplay among HIV, inflammation, traditional risk factors for cardiovascular disease, the adverse effects of antiretroviral therapy, and co-infections that may contribute to end-organ complications.12 Endothelial dysfunction is the most plausible link between HIV infection and atherosclerosis. Increased expression of adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and endothelial adhesion molecule (E-selectin), and inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin (IL-6), occur in HIV-positive patients. Higher plasma TNF-α, IL-6, and von Willebrand factor concentrations also correlate with viral load, thus suggesting an endothelial response to injury.13 Endothelial dysfunction may also occur after percutaneous coronary interventions in these patients, in whom restenosis rates may be higher than rates in other populations.14 Protease inhibitors are associated with dyslipidemia and insulin resistance.11,13 Amprenavir and fosamprenavir, with or without “boost” ritonavir or lopinavir with ritonavir, have the strongest association with MI; saquinavir and nelfinavir are not as clearly associated. The nucleoside reverse transcriptase inhibitors didanosine and abacavir are also associated with MI. Other agents, such as non-nucleoside reverse transcriptase inhibitors (nevirapine and efavirenz), entry inhibitors, and integrase inhibitors, do not appear to increase the risk for acute coronary events.11,15–17 Low-cholesterol diets during highly active antiretroviral therapy (HAART—generally three or more agents and usually a protease inhibitor) reduce the incidence of dyslipidemia.18 Exercise also greatly lowers lipid concentrations and helps prevent lipodystrophy.19 Lipids and glucose concentrations should be monitored.18,19 Patients with dyslipidemia should be managed by current guidelines for primary risk prevention and by avoiding known drug interactions, such as the interaction between simvastatin and ritonavir, which can increase simvastatin concentrations 400-fold.20,21 Premature cerebrovascular disease is also common in HIV-infected patients (see Chapter 59). The prevalence of stroke in AIDS patients was estimated to be 8% in a review of autopsies conducted between 1983 and 1987. Of 13 patients with stroke, 4 had cerebral emboli, and in 3 of them the embolus had a clear cardiac source. Investigation of acute stroke in HIV-infected individuals differs from that in the general population because causes include opportunistic infections, tumors, infectious and immune-mediated vasculopathy, and cardioembolism.20 Protease inhibitors markedly alter lipid metabolism and can be associated with premature atherosclerotic disease. Atherosclerotic disease in HIV-infected individuals is believed to be multifactorial in cause and prone to plaque rupture.13 In a large prospective study, the adjusted risk for MI was 16% per year of protease inhibitor exposure, almost double the rate of MIs over a 5-year period. The adjusted risk during treatment with non-nucleoside reverse transcriptase inhibitors increased by 5% per year, but significantly so. The risk for MI associated with protease inhibitors was attenuated when traditional risk factors were added to the model, which suggests that some, but not all the protease inhibitor–associated risk for MI can be attributed to metabolic changes.13 Protease inhibitors, specifically HAART overall, however, have decidedly reduced morbidity and mortality without short-term evidence of increased cardiovascular mortality.13 Lipodystrophy, including fat redistribution with increased truncal obesity; temporal wasting; elevated triglyceride concentrations; elevated concentrations of small, dense, low-density lipoprotein; and glucose intolerance should be treated because they increase the 10-year risk for cardiovascular events.8,14 Risk stratification based on traditional risk factors plus diet, alcohol intake, physical exercise, hypertriglyceridemia, cocaine or heroin use, thyroid disease, and hypogonadism should be considered for long-term prevention (Fig. 70-1 and Fig. e70-1 Fat redistribution is seen in 42% of children after more than 5 years of antiretroviral therapy.8 HIV is a moderate cardiovascular risk factor in children because early screening for lipid and glucose concentrations when starting antiretroviral therapy increasingly shows evidence of vascular dysfunction. Statins are reserved for those with very high cholesterol or triglyceride concentrations.12 The 2- to 5-year incidence of symptomatic heart failure ranged from 4% to 28% in HIV patients treated in the pre-HAART era, thus suggesting a prevalence of symptomatic HIV-related heart failure of between 4 and 5 million cases worldwide early in the epidemic.4,5 Although HAART has markedly reduced the incidence of clinically important systolic heart failure, it is available to only some of those in need.5,22,23 Asymptomatic cardiac structural abnormalities detected with echocardiography are also common and persist in patients receiving HAART: 18% have LV systolic dysfunction, 6.5% have LV hypertrophy, and 40% have left atrial dilation.24 LV systolic dysfunction is associated with a history of MI, elevated highly sensitive C-reactive protein concentrations, and current tobacco smoking.24 Among HIV-infected children up to 10 years old in the pre-HAART era, 25% died of chronic cardiac disease4,5 and 28% experienced serious cardiac events after an AIDS-defining illness.1,4,5,23 Importantly, even mildly decreased LV systolic function or increased LV mass in children is associated with increased mortality.25 Data from the HAART era suggest a markedly lower incidence of both structural abnormalities and clinical cardiomyopathy and a clear cardioprotective effect of HAART in children and adolescents.23,25–28 In HIV-infected patients, concurrent pulmonary infections, pulmonary hypertension, anemia, portal hypertension, malnutrition, or malignancy can alter or confuse the characteristic signs that define heart failure in other populations. Thus patients with LV systolic dysfunction can be asymptomatic or have New York Heart Association Class III or IV heart failure.21 Echocardiography (see Chapter 14) is useful for assessing LV systolic function in these patients and, in addition to diagnosing LV dysfunction, often reveals either low to normal wall thickness or LV hypertrophy and dilation, as well as left atrial dilation.4,11,21,24,25 Echocardiography should be performed at baseline and every 1 to 2 years thereafter or as clinically indicated in patients at elevated cardiovascular risk, with any clinical manifestations of cardiovascular disease, or with unexplained or persistent pulmonary symptoms or viral co-infections.4,10,21,24 Electrocardiography (see Chapter 12) can reveal nonspecific conduction defects and changes in repolarization. Chest radiography has low sensitivity and specificity for diagnosing heart failure in patients with HIV infection.25 In small studies of HIV-infected patients and in large populations of patients without HIV infection, blood brain natriuretic peptide concentrations have been inversely correlated with the LV ejection fraction and can be useful in the differential diagnosis of congestive cardiomyopathy in HIV-infected patients.11,29,30 Patients with encephalopathy are more likely to die of heart failure than are those without encephalopathy (hazard ratio, 3.4).4,21 HIV persists in reservoir cells in the myocardium and the cerebral cortex, even after antiretroviral therapy. Reservoir cells may hold HIV on their surfaces for extended periods and cause progressive tissue damage by chronic release of cytotoxic cytokines. Progressive LV dilation is common in HIV-infected children, in whom it may precede heart failure (5-year cumulative incidence, 12.3%) and is associated with increased LV mass, elevated LV afterload, and reduced LV function.23 In children vertically infected with HIV, early treatment with HAART for at least 5 years preserved cardiac structure and function and prevented heart failure better than in earlier groups, thus suggesting that HAART is protective.23 Several agents may cause HIV-related cardiomyopathy (see Table 70-1), including MI with HIV itself, tissue damage from myocarditis, drug-induced cardiotoxicities, effects of viral proteins, comorbid opportunistic infections, autoimmune responses to viral infection, nutritional deficiencies, and cytokine overexpression.21,23,28 Dilated cardiomyopathy can be related to the direct action of HIV on myocardial tissue or to proteolytic enzymes or cytokine mediators induced by HIV alone or in conjunction with co-infecting viruses (see Chapter 67).27 Toxoplasma gondii, group B coxsackievirus, Epstein-Barr virus, cytomegalovirus, adenovirus, and HIV in myocytes have appeared in endomyocardial biopsy specimens. Autopsy and biopsy findings have identified only scant, patchy inflammatory cell infiltrates in the myocardium.4,21,
Cardiovascular Abnormalities in HIV-Infected Individuals
Background
 ).3 Even more prevalent are treatment-related drug effects and interactions that directly challenge the cardiovascular system, such as lipid abnormalities with protease inhibitors and increased statin serum concentrations with protease inhibitors.6 Many drugs may prolong the QT interval or change repolarization, thereby increasing the risk for sudden cardiac death (see Chapters 9 and 37).6
).3 Even more prevalent are treatment-related drug effects and interactions that directly challenge the cardiovascular system, such as lipid abnormalities with protease inhibitors and increased statin serum concentrations with protease inhibitors.6 Many drugs may prolong the QT interval or change repolarization, thereby increasing the risk for sudden cardiac death (see Chapters 9 and 37).6
Accelerated Atherosclerosis
 ).13,14,18
).13,14,18
Left Ventricular Systolic Dysfunction
Incidence
Clinical Features
Pathogenesis
Myocarditis
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cardiovascular Abnormalities in HIV-Infected Individuals
70