Cardiac Tumors
Gerald Ross Marx
Adrian M. Moran
History of Cardiac Tumors
Columbus, a noted pathologist, recorded the first description of a cardiac tumor in 1562 (1). Subsequently, the first report of a cardiac tumor diagnosed in a living patient occurred in 1934, when the presence of myocardial involvement was inferred in a patient with metastatic disease and an abnormal electrocardiogram (ECG) (2). In 1942, surgical resection of two cardiac tumors was reported; both operations culminated in perioperative deaths (3,4). The first successful excision of a cardiac tumor, an atrial myxoma, was performed in 1955 (5).
Despite this rather long history, primary cardiac tumors were, and continue to be, quite rare, especially in the pediatric age group. Their atypical clinical presentation had often prevented timely diagnosis, with discovery at autopsy (6,7,8,9). Recent advances in noninvasive diagnosis and surgical management have resulted in marked improvement in recognition, survival, and long-term prognosis.
Most primary cardiac tumors in the pediatric age group are benign. These benign primary cardiac tumors maintain the potential for serious illness related to their conspicuous location. Furthermore, certain primary cardiac tumors can be associated with generalized systemic illness or peripheral emboli. Fewer than 10% of these primary tumors are malignant (6,7,9). Secondary tumors are also infrequent (7,9). Secondary tumor involvement continues to be quite rare in the pediatric age group although it is encountered more commonly than primary tumors in adults. Secondary tumors usually involve both the myocardium and pericardium. Primary pericardial tumors are also uncommon but again are important because early diagnosis and surgical intervention may be lifesaving.
Incidence
The incidence of cardiac tumors has been difficult to ascertain. Prior data have been limited to autopsy series, case reports, and reviews from large pediatric centers. Based on general autopsy series from patients of all ages, cardiac tumors had a reported incidence from 0.002% to 0.03% (9,10). Autopsy studies in children reported an incidence from 0.027% to 0.08% (6). The earliest reports, with the advent of echocardiography, reported an incidence of primary cardiac tumors of 0.0017% to 0.003%, using the number of hospital admissions as the common denominator (11,12). Using a similar database in a more recent era, 1980 to 1995, the reported incidence of primary cardiac tumors in fetuses to patients 18 years of age evaluated for heart disease at a large pediatric center had increased to 0.2% (13). The incidence of primary cardiac tumors, using the number of echocardiograms at a single pediatric institution from 1981 to 1997 as the denominator, had an incidence of 0.08% (14). In a seven-center collaborative study of 14,000 fetal echocardiograms, the incidence of primary cardiac tumors was 0.05% (15).
In an attempt to evaluate the incidence of primary cardiac tumors in pediatric patients, a retrospective review was performed between 1980 and 1998 using a computerized cardiology database at Boston Children’s Hospital. The incidence was based on a review of all first-time echocardiographic diagnoses of primary cardiac tumors in pediatric patients, which included fetal studies, newborns, infants, children, and adolescents. The denominator of the incidence was all first-time echocardiograms performed during the same time period. The rationale for choosing an echocardiography database implied that most, if not all, pediatric patients with significant murmurs, hemodynamic compromise, dysrhythmias, and associated systemic emboli or unexplained systemic illness seen at Boston Children’s Hospital would have undergone echocardiographic evaluation. The computerized pathology registry databases recorded during the same time interval were searched to determine whether any patients had had surgery or whether there had been a sudden death or incidental findings of a primary cardiac tumor that were not included in the echocardiography database. Only one additional patient was found. Hence, using the echocardiography database, 67 primary cardiac tumors were found in review of 38,952 studies, for an incidence of 0.17%. This is similar to the incidence of 0.2% reported in a similar manner during a concurrent time period at the Hospital for Sick Children, Toronto, Ontario (13). The incidence of 0.17% was also similar to 0.14% (15) reported in a multicenter review of fetal echocardiograms. Despite some of the inherent difficulties with these computations, this incidence does present interesting and important new information. The incidence of primary cardiac tumors in the pediatric age group appears to have increased nearly 10-fold. Moreover, this increased incidence is based on echocardiographic recognition of tumors in living patients, not on autopsy findings. This new incidence suggests that one or two new primary cardiac tumors will be detected for every 1,000 first-time pediatric echocardiograms.
Previous autopsy reports have reported that the most common primary cardiac tumors were rhabdomyomas (45%), fibromas (25%), myxomas (10%), intrapericardial teratomas (10%), and hemangiomas (5%) (7,9). The most common tumors in newborns and infants were rhabdomyomas, fibromas, and intrapericardial teratomas. In older children and adolescents, myxomas, rhabdomyomas, and fibromas predominated. Using the recent computerized databases of echocardiography, surgery, pathology, catheterization, magnetic resonance imaging (MRI), and computerized axial tomography, the frequencies of primary cardiac tumors were evaluated at Boston Children’s Hospital from 1980 to 2005 (Table 72.1). A total of 129 tumors was found with frequencies as follows: rhabdomyomas (60.5%), myxomas (13.9%), fibromas (7.8%), pericardial teratomas (1.6%), others (7.0%), nonspecified (5.4%), and metastatic dissemination (3.9%) (Table 72.1). “Others” included three hemangiomas, a mass related to excessive eustachian valve tissue, cardiac calcification, blood cyst of the tricuspid valve, neurofibroma in a patient with neurofibromatosis, hypereosinophilia in a patient with Loeffler syndrome, and one Purkinje cell tumor. Five patients had metastatic tumor dissemination to the heart. Their primary diagnoses included rhabdomyosarcoma, hepatoblastoma, Ewing sarcoma, testicular carcinoma, and malignant melanoma. Three of the five presented with symptoms related to tumor pulmonary emboli. The increased relative incidence of rhabdomyomas seems related to the increased diagnosis of tuberous sclerosis and surveillance echocardiography in patients with known or suspected tuberous sclerosis at a specialized tertiary medical center.
TABLE 72.1 Frequencies of Cardiac Tumors at Boston Children’s Hospital From 1980 to 2005 | |||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||
In this Boston Children’s hospital experience, in patients diagnosed younger than 1 month of age (including fetal studies), rhabdomyomas were most often diagnosed (80%), followed by myxomas (5%), pericardial teratomas (5.0%), fibromas (2.5%), others (5.0%), and nonspecified (2.5%) (Table 72.1). All seven of the tumors diagnosed in utero were rhabdomyomas, and six of these patients were subsequently diagnosed with tuberous sclerosis. In a seven-center retrospective review of 14,000 fetal echocardiograms, 19 patients had tumors for a fetal incidence of 0.14% (15). Seventeen of the 19 (89%) were rhabdomyomas and 10 (54%) had tuberous sclerosis. The other tumors included a fibroma and atrial hemangioma. In a similar multicenter retrospective review of patients diagnosed either in utero or at younger than 1 month of age, 84 of 94 patients with cardiac tumors had rhabdomyomas (89%); 80% of these patients had tuberous sclerosis (16).
In patients older than 1 year of age diagnosed at Boston Children’s Hospital, rhabdomyomas were diagnosed in 45%, myxomas 23%, fibromas 8%, other 9%, nonspecified 7%, and metastatic dissemination 7% (Table 72.1). The ages of those patients diagnosed with metastatic tumor involvement ranged from 6 to 15 years of age. The median age of presentation for all myxomas was 7 years. The relative increased incidence of myxomas in recent years seems in part attributable to earlier referral and subsequent echocardiographic diagnosis. The increased detection of secondary tumor involvement in the pediatric age group most probably relates to increased awareness and early referral for echocardiographic evaluation.
Diagnostic Procedures
Electrocardiogram and Chest Roentgenogram
Early studies reported abnormalities on chest roentgenograms in 80% of patients with cardiac tumors (17). Findings including cardiomegaly, abnormal cardiac silhouette, and calcification are nonspecific. Only 47% of patients with primary cardiac tumors have abnormal ECGs. However, in patients with known tumor involvement, the ECG can show ST segment abnormalities or strain, and ventricular hypertrophy. Additionally, dysrhythmias may be the presenting mode of presentation of cardiac tumors in pediatric patients of all ages. Some have ECG evidence of pre-excitation.
Echocardiography
More than two decades of collective experience have established 2-D Doppler echocardiography as the primary diagnostic procedure for the evaluation of cardiac tumors in pediatric patients (11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44). Two-dimensional Doppler echocardiography is expedient, noninvasive, and accurate. The anatomic and hemodynamic abnormalities can be determined quickly, and critically ill patients can be sent for surgical intervention. More stable patients can be monitored serially by using this noninvasive technique. Echocardiography is also useful for the follow-up of patients after surgical removal of the tumor (45). Two-dimensional echocardiography may aid in tumor differentiation (46,47). Strain and strain rate Doppler tissue imaging has been applied to differentiate tumors based on inherent properties of tissue deformation (48). Transesophageal echocardiography (TEE) provides precise delineation of cardiac tumors and can be used intraoperatively to guide surgical management (49,50,51,52,53,54,55). In adults, TEE has been reported to provide improved determination of tumor attachment sites as well as the extent of myocardial/pericardial involvement (55).
Recent developments in matrix array 3-D echocardiography allow timely analysis of tumor volume (Fig. 72.1A, B–C). The spatial relationship of tumors to other cardiac structures, including atrioventricular and semilunar valves, and inflow and outflow tracts can readily be appreciated with 3-D echocardiography (56,57).
Magnetic Resonance Imaging
MRI also shows the location, number, and size of cardiac tumors in the pediatric age group (29,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73). MRI is also important in the clinical management of secondary tissues because it can visualize mediastinal and other intrathoracic sites (65). Spin echo MR images provide high-resolution images to enhance delineation of the specific pathology of intracavitary, valvar, myocardial, pericardial, and juxtacardiac masses (Fig. 72.2A). Fast gradient cine MR provides dynamic imaging techniques to provide additional hemodynamic
information (66). MRI can be particularly helpful in visualization of spatial relation of the tumor mass to the coronary arteries, which has been helpful in guiding surgical management. In comparative studies, 2-D echocardiography appears to have greater sensitivity than MRI for the detection of intramural (29) and intracavitary cardiac tumors (60); however, MRI has been superior in detection of apical tumors (60). MRI has inherent advantages over echocardiography including wider field of view, negligible interference by bone or lung tissue (58) and has the ability to differentiate tissue type (74). Limitations of MRI include requirement for sedation in young patients, to eliminate patient motion and to control respiration frequently with the use of general anesthesia (29,59). Dysrhythmias caused by the tumor can limit ECG gating, which can result in data misregistration and consequently poor image quality (29).
information (66). MRI can be particularly helpful in visualization of spatial relation of the tumor mass to the coronary arteries, which has been helpful in guiding surgical management. In comparative studies, 2-D echocardiography appears to have greater sensitivity than MRI for the detection of intramural (29) and intracavitary cardiac tumors (60); however, MRI has been superior in detection of apical tumors (60). MRI has inherent advantages over echocardiography including wider field of view, negligible interference by bone or lung tissue (58) and has the ability to differentiate tissue type (74). Limitations of MRI include requirement for sedation in young patients, to eliminate patient motion and to control respiration frequently with the use of general anesthesia (29,59). Dysrhythmias caused by the tumor can limit ECG gating, which can result in data misregistration and consequently poor image quality (29).
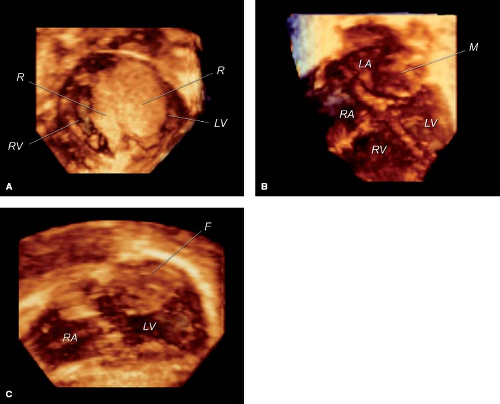 Figure 72.1 Three-dimensional echocardiograms using a matrix array transducer. A: Large right and left ventricular rhabdomyomas (R) in a newborn. B: Round, multilobulated left atrial myxoma (M) in a 2-year-old child. C: Large left ventricular fibroma (F) in a 4-year-old boy. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
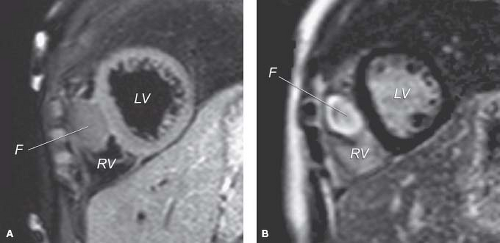 Figure 72.2 A: Spin-echo MRI from a child with a large right ventricular fibroma (F). B: Gadolinium-enhanced MRI, from same child, demonstrating uptake within tumor mass. This uptake indicates fibrous tissue, consistent with a cardiac fibroma. LV, left ventricle; RV, right ventricle. (Courtesy of Tal Geva, MD, Department of Cardiology, Children’s Hospital, Harvard Medical School, Boston, MA.) |
MRI assessment allows differentiation of tumor from myocardium (68) and differentiation of the tumor type (Fig. 72.2B) (69,70,71,72,73). In a recent multicenter experience of several pediatric centers, 97% of the tumor cases were correctly diagnosed by comprehensive MRI examinations including a differential diagnosis in 42%.
Employing a variety of imaging techniques, MRI can help to differentiate tumor type (Table 72.2). Fast gradient cine imaging (typically white blood sequences using a Steady State Free Precession [SSFP]) provides anatomic and physiologic information (66). Spin echo black blood images provide high-resolution anatomic and tissue characterization information (e.g., fat is bright [hyperintense] on T1-weighted sequences, while fluid/edema is bright on a T2-weighted sequence). Using gadolinium-based sequences (first-pass perfusion imaging and myocardial delayed enhancement sequences), vascularity and fibrosis of tumors can be assessed. For example, fibromas display a hyperintense signal (as compared to myocardium) and have distinct enhancement with late gadolinium imaging due to tumor fibrosis (Table 72.2). Hemangiomas have intense enhancement with first-pass gadolinium due to the vascularity, and no or mild intensity with late gadolinium. Rhabdomyomas have isointense typical T1 and T2 imaging, and no distinct increased intensity with first- or second-pass gadolinium imaging (75).
The differentiation of tumor type is essential in the care of patients. For example, rhabdomyomas may regress in size without intervention, or may respond to treatment with specific inhibitors of cell growth and differentiation, fibromas may enlarge, and recent evidence suggests that hemangiomas may resolve with steroid or interferon administration.
Angiography
Invasive angiography also has been used to diagnose cardiac tumors. This modality provides indirect and nonspecific findings of intracavitary filling defects or cavity obliteration by large intramural tumors. Pressure measurements can be obtained with catheterization, with the inherent risk of placing a catheter near or across a friable tumor. Tissue diagnosis can be attempted by biopsy technique, with the risk of embolizing tumor fragments (44).
Rhabdomyomas
Rhabdomyomas constitute 45% to 80% of all primary cardiac tumors in the pediatric age group (7,9,10,11,12,13,14) (Table 72.1; Videos 72.1 to 72.6). They are considered the most common primary cardiac tumor in this age group (7,9,10,11,12,13,14). These tumors can be diagnosed in the prenatal period but are most frequently diagnosed in the newborn infant (74,76,77,78,79,80,81). Occurrences of sudden death in pediatric patients of all ages, including stillbirths, have been attributed to cardiac rhabdomyomas (8,76,77,78,79,80,81,82).
TABLE 72.2 MRI Can Help to Differentiate Tumor Type | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In general, rhabdomyomas are multiple, well-circumscribed, noncapsulated, white or gray-white intramural or intracavitary nodules that can occur anywhere within the heart (8,9,21,22,25,76,79,80,81), most commonly involving the ventricles (83). Although intramural, these large tumors can encroach on the intracavitary space. Other tumors occur as intracavitary pedunculated masses, or attached by a broad base to the endocardial surface (8,82,83,84). Cardiac rhabdomyomas occur as single intramural or intracavitary
masses in 10% of patients (8,9,85). Histologically, rhabdomyomas contain large vacuolated cells filled with glycogen (Fig. 72.3). Typical spider cells are seen with eccentric nuclei, granular cytoplasm, and thin cytoplasmic extensions projecting toward the cell membrane (8). Rhabdomyomas often are classified as hamartomas, with an inability of cells to undergo mitotic division (8,86,87).
masses in 10% of patients (8,9,85). Histologically, rhabdomyomas contain large vacuolated cells filled with glycogen (Fig. 72.3). Typical spider cells are seen with eccentric nuclei, granular cytoplasm, and thin cytoplasmic extensions projecting toward the cell membrane (8). Rhabdomyomas often are classified as hamartomas, with an inability of cells to undergo mitotic division (8,86,87).
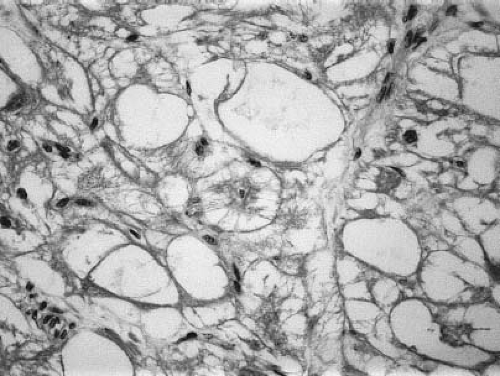 Figure 72.3 Rhabdomyoma. Photomicrograph showing cells with clear cytoplasm, vacuolization, and an occasional spider cell (center). (Hematoxylin and eosin stain.) (Courtesy of Peter Faul, MB, BCh, Department of Pathology, Children’s Hospital, Harvard Medical School, Boston, MA.) |
A rare form of cardiomyopathy, rhabdomyositis, has been described. Tumor nodules are not grossly apparent; microscopically, however, the cardiac muscle fibers and conduction system are diffusely involved with rhabdomyomatous histologic changes (88,89). Recurrent atrial tachycardia and sudden death from intractable ventricular tachycardia have been attributed to rhabdomyositis (88,89).
The clinical findings of cardiac rhabdomyomas are related to the number, position, and size of the tumors. Large intramural or intracavitary rhabdomyomas may obstruct the intracavitary space or the orifice of the atrioventricular or semilunar valves (22,74,76,77,78,79). Extensive cardiac involvement has been associated with diminished myocardial function (8,19,42,74,78). Direct compression of the conduction system can result in serious dysrhythmias (8,28,38,65,74,76,77,78,79,80,81,82,83,84,85). Rhabdomyomas have been diagnosed by 2-D echocardiography in the fetus (Fig. 72.4) (28,77,90,91,92,93,94,95,96). Such prenatal detection has been made while screening fetal dysrhythmias, nonimmune hydrops, decreased fetal growth, and familial tuberous sclerosis (28,77,78,79,80,81,90,91,92,93,94,95,96). Postnatally, patients may present without obvious clinical findings, despite extensive cardiac involvement (6,7,41,74,85). Others may have only a murmur of valvular obstructive disease (42,76,79).
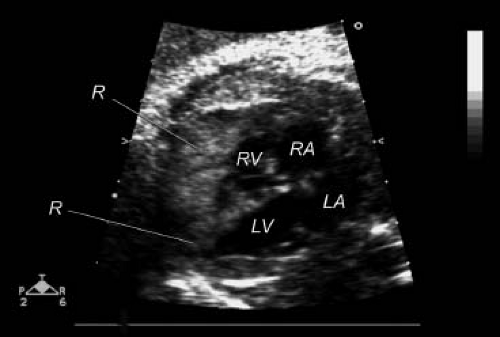 Figure 72.4 Two-dimensional fetal echocardiogram at 22 weeks’ gestation, demonstrating right and left ventricular rhabdomyomas (R). In utero, a brain MRI scan showed intracranial masses, and at birth the baby was diagnosed as having tuberous sclerosis. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
Newborns and infants with large rhabdomyomas are often critically ill and present with respiratory distress, congestive heart failure, and low cardiac output (8,19,74,76,77,78,79,80,81,82). Rhabdomyomas may simulate hypoplastic left heart syndrome when tumors obliterate the left ventricular cavity and severely obstruct blood flow across the mitral or aortic valves (74,97,98,99). Cardiomyopathy has been diagnosed in newborns and infants with extensive myocardial infiltration (19,42,79). Single pedunculated tumors have been associated with subaortic stenosis (40,42,98,99,100).
Right atrial or right ventricular rhabdomyomas can cause low cardiac output and signs and symptoms of right-sided heart failure (76). Extreme cyanosis can occur when tumors obstruct the tricuspid valve or right ventricular outflow tract, resulting in right-to-left atrial shunting (21,40,42,76,79). Single obstructive pedunculated rhabdomyomas may simulate tricuspid atresia, tetralogy of Fallot, or critical valvar pulmonic stenosis of the newborn (84). The clinical presentation may be even more confusing when rhabdomyomas coexist with significant congenital heart defects (101,102).
Abnormal ECG findings include left axis deviation, atrial enlargement, ventricular hypertrophy, and ST-T wave abnormalities consistent with ischemia and/or strain (19,74,76,79). These abnormal ECG findings are also indicative of extensive intramural infiltration. Involvement of the conduction system can be inferred from baseline ECGs that manifest bundle branch block (74,76,79), pre-excitation (26,38,40,74,79), or first- through third-degree atrioventricular block (6,28,74,76,77,79,82). Sudden death has been attributed to arrhythmias in pediatric patients of all ages (8,19,20,28,76,77,78,79,80,81,82). These arrhythmias may be a result of either severe hemodynamic compromise or contiguous location of tumors to the conduction system. All major rhythm disturbances have been reported, including sinus bradycardia, atrial and ventricular tachycardias, and first- through third-degree atrioventricular block. In a more recent large review of 106 patients with rhabdomyomas, 17 (16%) had significant arrhythmias (103). Six percent had ventricular tachycardia and 10% had manifest pre-excitation. In most cases, the rhythm disturbances seemed to resolve with natural tumor regression. When necessary, surgical excision and/or radiofrequency catheter ablation therapy was effective (103).
The chest radiograph may appear normal in older patients with cardiac rhabdomyomas. Occasionally, the only abnormal finding
may be distortion of the cardiac silhouette, especially when the rhabdomyomas extend to the epicardium. The chest radiograph, however, often shows cardiomegaly and pulmonary edema in the critically ill neonate and infant (7,19,40,74,76,77,78,79).
may be distortion of the cardiac silhouette, especially when the rhabdomyomas extend to the epicardium. The chest radiograph, however, often shows cardiomegaly and pulmonary edema in the critically ill neonate and infant (7,19,40,74,76,77,78,79).
The 2-D and 3-D echocardiographic characteristics of cardiac rhabdomyomas are highly echogenic, multiple, well-circumscribed intramural or intracavitary nodules occurring anywhere within the heart (Figs. 72.1A and 72.5) (22,38,41,47,85,89,90,91,92,103). Rhabdomyomas also may be visualized as either single pedunculated intracavitary or intramural masses (23,40,41,42,78,85,97,103). Rhabdomyomas have a homogeneous, echo-bright, finely speckled pattern (103). In contrast, intracardiac thrombi, myxomas, and hemangiomas have circumscribed echolucent areas as a result of hemorrhage formation. Moreover, rhabdomyomas do not have interspersed, distinct, echogenic regions consistent with calcification or fibrosis. Rhabdomyomas rarely have an associated pericardial effusion (21). Fetal rhabdomyomas are readily diagnosed in utero (Fig. 72.4), and in recent multicenter studies, the in utero incidence of rhabdomyomas was as high as 88% (15) and 89% (16), respectively. Multiple tumors are highly indicative of rhabdomyomas both in the fetus and neonate (15,16,80,81). Fetal rhabdomyomas have been reported to grow in utero ≤32 weeks’ gestation and then regress in size (81).
Cardiac rhabdomyomas are closely associated with the syndrome of tuberous sclerosis. Tuberous sclerosis is a complex disorder that is transmitted as an autosomal dominant trait of variable expression (104,105,106,107,108,109,110,111,112,113). The incidence of tuberous sclerosis has been reported to be 1 in 6,000; two-thirds of cases are sporadic resulting from mutation (81). In an autopsy series, 31% of patients with tuberous sclerosis were reported to have had rhabdomyomas (8). Fifty percent of patients with tuberous sclerosis had echocardiographic evidence of tumor involvement (15,85). More recent studies indicate a higher incidence of patients with rhabdomyomas having tuberous sclerosis. Fetal studies indicate that 52% to 79% of all fetuses with cardiac tumors have tuberous sclerosis (15,16,80). The incidence of tuberous sclerosis is as high as 59% to 80% in patients with confirmed fetal rhabdomyomas. Although not invariably, multiple rhabdomyomas are more consistent with the diagnosis of tuberous sclerosis than a singular tumor, with one report indicating that in the fetus or newborn with multiple ventricular tumors, 95% had tuberous sclerosis (16). However, in the latter report, 23% of fetuses or neonates with a singular tumor had tuberous sclerosis as well. In the fetus and neonate, MRI imaging can demonstrate the association of cerebral nodules, establishing the diagnosis of tuberous sclerosis in these patients. The clinical importance of the association between tuberous sclerosis and cardiac rhabdomyomas is evident in a report indicating that 1 in 40 patients with tuberous sclerosis may die as a direct result of cardiac rhabdomyomas (106).
In addition to cardiac involvement, tuberous sclerosis can affect almost all organ systems, including the brain, kidney, pancreas, retina, and skin (104,105,107,108,109). Therefore, clinical manifestations of tuberous sclerosis may not be clinically apparent in mildly affected patients (107,108,112). Even in severely affected patients, clinical manifestations such as Shagreen patches, adenoma sebaceum, seizures, and mental retardation may not become evident until later in life (105,107,108,109,110,111,112). Recently cardiac fat containing lesions have been reported in adults with tuberous sclerosis which are distinct from rhabdomyomas and are seen in one third of adolescent/ adult patients. There appears to be a higher incidence of abdominal angiomyolipomas in such patients. The clinical significance of these lesions requires further study (114). Despite the variability in clinical presentation, a family history of tuberous sclerosis or evidence of other organ system involvement may aid in establishing the diagnosis. This is especially germane to the neonate who presents with an intracardiac mass. Neonates with tuberous sclerosis, however, may have no manifestations of this syndrome other than cardiac tumors (79,90,94,95,108). Furthermore, a negative family history does not preclude the diagnosis since ≤50% of cases of tuberous sclerosis may be spontaneous mutations. Recent advances in genetic testing may elucidate more fully the inheritance of tuberous sclerosis. To date, two loci associated with tuberous sclerosis have been identified on chromosome 9q34 (TSC1-hamartin gene) and 16p13.3 (TSC2-tuberin gene) (111,112). These loci code for proteins, hamartin and tuberin, that have a tumor-suppressor function (113).
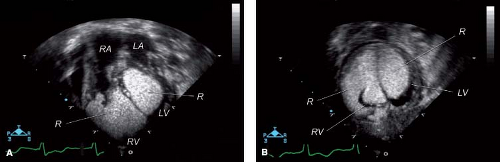 Figure 72.5 Two-dimensional echocardiogram in a newborn infant demonstrating very large right and left ventricular rhabdomyomas (R). The patient was diagnosed with tuberous sclerosis. Despite the large tumors, surgery was not undertaken, and the tumors regressed by 2 years of age. A: Four-chamber view showing extensive right and left ventricular tumors. B: Orthogonal short-axis views demonstrating the extensive tumor mass. Ao, aorta; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
Prostaglandin E1 (PGE1), by maintaining patency of the ductus arteriosus, may help stabilize critically ill newborns in whom cardiac rhabdomyomas cause severe right or left ventricular obstruction. Prompt surgical excision is indicated for life-threatening hemodynamic compromise or arrhythmias (38,39,76,79) and is required in ≤23% of patient series (97). Partial excision may provide significant relief of inflow or outflow tract obstruction when attempts at complete resection would severely damage the remaining myocardium
(19,21,42). The Ross procedure has been proposed when the aortic valve is severely compromised (115). Surgical intervention may be highly successful, without compromising either myocardial or valvular function, in patients with single intracavitary rhabdomyomas (40,84,100). Medical treatment has been successful for the treatment of severe dysrhythmias, especially as the tumor resolves. Others have reported successful radiofrequency ablation therapy in selected patients with rhabdomyomas and supraventricular tachycardia (116). However, even very large rhabdomyomas may significantly regress in size or disappear completely without intervention (22,38,39,90,100). Unoperated patients may have minimal cardiac signs or symptoms several years later (90). Therefore, the presence of rhabdomyomas alone, without life-threatening hemodynamic instability or arrhythmias, should not be an absolute indication for surgery (18,19,38,39,90,100). Recent case studies have reported successful treatment of life-threatening cardiac rhabdomyomas in newborns infants with mTOR (mammalian target of rapamycin) inhibitors (117,118,119,120). The TSC1 and TSC2 genes code for hamartin and tuberin, which form a tumor suppressor heterodimer that inhibits the protein (serine/threonine) kinase mTOR. When upregulated, mTOR leads to abnormal cell proliferation and growth (120) and the formation of harmatomatous lesions. The mTOR inhibitors have been reported to cause successful regression of multiple tumors associate with right and left ventricular outflow tract obstruction (117,118,119), and resolution of life-threatening dysrhythmias (120) caused by multiple nonobstructive intramural tumors.
(19,21,42). The Ross procedure has been proposed when the aortic valve is severely compromised (115). Surgical intervention may be highly successful, without compromising either myocardial or valvular function, in patients with single intracavitary rhabdomyomas (40,84,100). Medical treatment has been successful for the treatment of severe dysrhythmias, especially as the tumor resolves. Others have reported successful radiofrequency ablation therapy in selected patients with rhabdomyomas and supraventricular tachycardia (116). However, even very large rhabdomyomas may significantly regress in size or disappear completely without intervention (22,38,39,90,100). Unoperated patients may have minimal cardiac signs or symptoms several years later (90). Therefore, the presence of rhabdomyomas alone, without life-threatening hemodynamic instability or arrhythmias, should not be an absolute indication for surgery (18,19,38,39,90,100). Recent case studies have reported successful treatment of life-threatening cardiac rhabdomyomas in newborns infants with mTOR (mammalian target of rapamycin) inhibitors (117,118,119,120). The TSC1 and TSC2 genes code for hamartin and tuberin, which form a tumor suppressor heterodimer that inhibits the protein (serine/threonine) kinase mTOR. When upregulated, mTOR leads to abnormal cell proliferation and growth (120) and the formation of harmatomatous lesions. The mTOR inhibitors have been reported to cause successful regression of multiple tumors associate with right and left ventricular outflow tract obstruction (117,118,119), and resolution of life-threatening dysrhythmias (120) caused by multiple nonobstructive intramural tumors.
Fibromas
Fibromas are generally reported as the second most common primary cardiac tumor in the pediatric age group (Videos 72.7 to 72.10) (1,7,9,13). However, in a more recent review of the Boston Children’s Hospital database from 1980 to 2005, fibromas were the third most common tumors (8%) (Table 72.1). Although fibromas have recently been reported in utero (14,15) and in patients younger than 1 month of age (34,35,36,37,121,122,123,124), they are found much less commonly than rhabdomyomas in this age group. In the recent review at Boston Children’s Hospital, fibromas were the second most common tumors (17%) in patients diagnosed between 1 month and 1 year of age (Table 72.1). These primary tumors are rarely seen in older children, adolescents, or young adults. Sudden death has been attributed to cardiac fibromas in pediatric patients of all ages (31,124,125). To date, no distinct genetic inheritance or familial predisposition has been associated with cardiac fibromas. These tumors have been associated with Gorlin syndrome, which includes multiple nevoid basal cell carcinomas, cysts of the jaw, and diffuse skeletal abnormalities (126). Cardiac fibromas also have a rare association with familial adenomatous polyposis and its subtype Gardner syndrome (127,128).
Cardiac fibromas are predominantly single, white, firm, nonencapsulated, intramural tumors that involve the left ventricular free wall or interventricular septum (32,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129). These tumors often are located at the left ventricular apex. Less frequently, fibromas can be multiple and invade the right ventricular free wall, atrial septum, or atrial free wall (32,35,36,37,123,124). Extensive intramural fibromas can encroach and obliterate the intracavitary space (35,36,37,123,124,129). Although rare, intracavitary fibromas have been reported (35,36,37,123,124,129), occurring either as a single pedunculated mass (37) or attached by a broad base to the endocardium (34). Both the mitral and tricuspid valve leaflets can be entangled within the tumor mass, causing significant valvar regurgitation (34,35). Similar to rhabdomyomas, fibromas also can be associated with congenital heart defects (91). Cardiac fibromas have been reported to increase in size both prenatally and postnatally (13,14). Histologically, cardiac fibromas consist of fibroblasts, collagen fibers, and minimal elastic tissue (Fig. 72.6) (32,33,35,121,124,129). Occasionally, calcium can be interspersed within the tumor (33,113).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


