Cardiac Channelopathies, Syncope, and Sudden Death
David J. Tester
Michael J. Ackerman
In the United States, an estimated 300,000 to 400,000 individuals die suddenly each year with the vast majority secondary to coronary artery disease in the elderly (1). Fewer than 10,000 of these deaths involve people <35 years of age. Sudden cardiac death (SCD) in children, adolescents, and young adults is infrequent with an incidence between 1.3 and 8.5 per 100,000 patient-years (2). Sudden death under the age of 1 year can be attributed to infection, cardiovascular anomalies, child abuse/negligence, accidents, homicide, or metabolic/genetic disorders. However, 70% to 80% of these infantile deaths have no identifiable cause and are labeled as sudden infant death syndrome (SIDS) (3,4). SIDS remains the leading cause of postneonatal infant death and the third leading cause of infant mortality overall in the United States with an estimated incidence of 0.57 per 1,000 live births (5,6).
Beyond the first year of life, the cause and manner of death can be established from a comprehensive medicolegal investigation that includes an autopsy (7,8). For nearly half of young victims from 1 to 39 years of age, there are no apparent warning signs and sudden death often occurs as the sentinel event. The postmortem investigation is critical to determine the cause and manner of death (2). A postmortem examination may detect a noncardiac cause of the death such as asthma, epilepsy, or pulmonary embolism. However, SCD is the prevailing cause of sudden death in the young, with structural cardiovascular abnormalities often evident at autopsy, including hypertrophic cardiomyopathy (HCM) (Chapter 52), arrhythmogenic right ventricular cardiomyopathy (ARVC)/arrhythmogenic cardiomyopathy (ACM) (Chapters 22, 38), congenital coronary artery anomalies (Chapter 32), and myocarditis (Chapter 56) (8,9).
In contrast, approximately 33% to 50% of sudden deaths involving previously healthy children, adolescents, and young adults, have no identifiable abnormalities at autopsy, and the death is classified as autopsy-negative sudden unexplained death (SUD) (7,8,10,11,12). The exact prevalence of SUD, particularly in children, is unclear. Potentially lethal and heritable channelopathies such as congenital long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS) leave no clues at autopsy. The absence of any material evidence should prompt coroners, medical examiners, and forensic pathologists to consider that a fatal arrhythmia might have caused the SUD (7,13,14,15,16). Accordingly, postmortem genetic testing (aka, the molecular autopsy) involving a targeted analysis of the channelopathy-susceptibility genes or an expanded analysis of the 100+ genes implicated in sudden death, and now increasingly, whole exome sequencing (WES) with its interrogation of ∼20,000+ genes, can elucidate potentially the pathogenic mechanism and establish probable cause and manner of SUD (17,18,19,20,21). Most often the arrhythmias associated with these cardiac channelopathies spontaneously return to normal sinus rhythm resulting in an episode of “just” syncope. As such, it is paramount to distinguish the ordinary benign faint from a sudden death warning sign.
In this chapter, we will summarize some recent population-based investigations of sudden death in the young to better understand its frequency and causes. Next, the pathologic bases, clinical evaluation and diagnosis, and therapeutic management of the cardiac channelopathies will be detailed. Finally, we will examine the features of fainting spells that may allow discrimination between common but benign vasovagal/neurocardiogenic syncope and much rarer but, potentially lethal, cardiac channelopathy-precipitated (arrhythmic) syncope.
The Prevalence and Causes of Sudden Death in the Young
In 1996, Maron et al. (8) demonstrated that HCM was the most frequent cause of SCD involving young competitive athletes. In this setting, over one-third of SCDs were attributed to HCM and an additional 10% had an unexplained increase in cardiac mass, representing “possible HCM” (Fig. 20.1A). Autopsy-negative SUD accounted for only 3% of cases. The preponderance of athletes were male (90%) and collapsed during or instantaneously following a daytime training session (90%). Accordingly, HCM may represent the most common cause of SCD on the athletic field in the United States. However, in a most recent study involving SCD among NCAA athletes, the most common cause of SCD was a structurally normal heart or autopsy-negative SUD in 34% of the decedents (Fig. 20.1B) (22).
In Italy, Corrado et al. (9) noted that ARVC/ACM overshadowed HCM as the most frequent cardiomyopathy associated with SCD in young Italians (Fig. 20.1C). In this autopsy review of 273 cases of Italian SCD, atherosclerotic coronary artery disease prevailed at 20% followed by ARVC (13%) and HCM (7%). Hearts were identified as structurally normal (SUD) in 6% of cases of SUD. In 2005, Puranik et al. reviewed the autopsy reports from 427 young victims of unexpected sudden death (ages 5 to 35 years) over a 10-year period in eastern Sydney, Australia (Fig. 20.1D) (23). This population-based cohort included ≥90% of all sudden deaths that occurred in this urban population during the study period. Excluded were traumatic, accidental, selected drownings, and death from drug intoxications. SCD was determined for more than half of these SUDs. Cardiomyopathies accounted for only 16% of their SCDs and only 6% of deaths were attributed to HCM. Instead, autopsy-negative SUD (29%) was the leading cause of SCD (Fig. 20.1D) with nearly 10% of the decedents with structurally normal hearts deemed to have LQTS.
Further, a 25-year review of autopsies performed on American military recruits by Eckart et al. showed a nontraumatic sudden death rate of 13 per 100,000 recruit years among a monitored 6.3 million men and women age 18 to 35 years (Fig. 20.1E) (23). Of the 126 sudden deaths, 108 (86%) were related to exercise. Half of the sudden deaths were attributed to an identifiable cardiac abnormality after a postmortem investigation. However, 35% of the sudden deaths were classified as autopsy-negative SUD (23). Several cases were identified as having a family history of sudden premature death suggesting a heritable predisposition for a malignant arrhythmia (23).
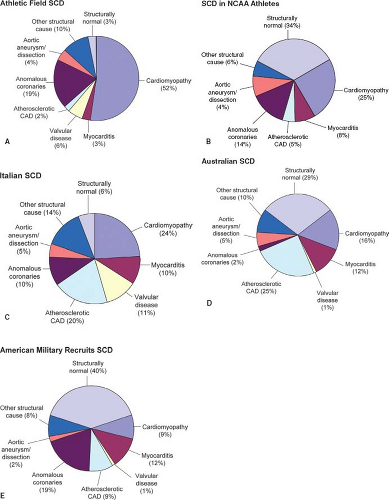 Figure 20.1 Epidemiology of sudden death in the young. A comparison of data from five cohorts. A: Maron et al. (1996) (8). N = 134; mean age: 17 years. Frequency of cardiomyopathy subtypes: hypertrophic cardiomyopathy (HCM) 36%, dilated cardiomyopathy (DCM) 3%, arrhythmogenic right ventricular dysplasia (ARVD) 3%, and unexplained increase in cardiac mass (“possible HCM”) 10%. B: Harmon et al. (2014) (22). N = 36; age range: 17 to 24 years. Frequency of cardiomyopathy subtypes: idiopathic left ventricular hypertrophy (LVH)/possible HCM 8%, HCM 3%, DCM 8%, and ARVD 3%. C: Corrado et al. (2001) (9). N = 273; mean age: 24 years. Frequency of cardiomyopathy subtypes: HCM 7%, DCM 4%, and ARVD 13%. A significant fraction of those included in “other structural causes” (24/38) had histologic evidence of conduction system abnormalities. D: Puranik et al. (2005) (12). N = 241, mean age: 27 years. Frequency of cardiomyopathy subtypes: HCM 6%, DCM 5%, ARVD 2%, and LVH 3%. E: Eckart et al. (2004) (23). N = 108, mean age: 19 years. Frequency of cardiomyopathy subtypes: HCM 8%, DCM 1%, ARVD 1%. “Structurally normal” includes the diagnosis of arrhythmia disorders, such as long QT syndrome, as well as all sudden unexplained deaths. In some instances, minimal structural abnormalities were noted at autopsy, but these were felt to be insufficient to cause sudden death. “Cardiomyopathy” includes those with HCM, DCM, ARVD, and those with unexplained increase in cardiac mass not meeting strict criteria for HCM (i.e., “possible HCM”). CAD, coronary artery disease. (From Tester DJ, Ackerman MJ. The role of molecular autopsy in unexplained sudden cardiac death. Curr Opin Cardiol. 2006;21(3):166–172 (24).) |
These population-based studies suggest that while the majority of SCD stem from identifiable morphologic abnormalities found at autopsy, one-third and perhaps as many as half of sudden deaths involving previously healthy children, adolescents, and young adults are autopsy-negative SUD.
Autopsy-Negative Sudden Unexplained Death
Assessment of Family Members
In cases of autopsy-negative SUD, a cardiac and genetic evaluation of first and second-degree relatives and/or a molecular autopsy may elucidate the underlying mechanism of the sudden death. Although arrhythmias can be isolated events, some may represent manifestations of inherited arrhythmia syndromes with increased risk for syncope, cardiac arrest, or SCD in relatives when there is a family history of sudden death. Thus, first-degree relatives of the decedent should have a comprehensive cardiovascular evaluation including an extensive personal and family history, physical examination, 12-lead electrocardiogram, treadmill stress test, 24-hour Holter monitoring, and an echocardiogram.
In 2003, Behr et al. performed a detailed cardiovascular evaluation of 109 first-degree relatives of 32 cases of SUD and showed that 22% of these families had evidence of inherited cardiac disease with the majority having features suggestive of LQTS (10). Similarly, in 2005, Tan et al. found that 28% of families had an identifiable cardiac channelopathy including CPVT and LQTS following a clinical assessment of first-degree relatives of young SUD victims (25). In a 2008 follow-up study by Behr et al., a diagnosis of inheritable heart disease was made in 53% of first-degree relatives of SUD victims following a comprehensive clinical evaluation; 70% being diagnosed with either LQTS (53%) or BrS (17%). Strikingly, 30% of the families evaluated reported a family history of additional unexplained premature sudden deaths under the age of 45 years, and nearly 20% of the decedents had a prior history of syncope.
In 2010, van der Werf et al. identified a certain or probable diagnosis in 47 (33%) of 140 families of SUD victims (aged 1 to 50 years) following a clinical cardiac assessment. Ninety-six percent of these 47 families were diagnosed with an inherited cardiac disease (21% LQTS, 17% CPVT, 15% BrS, and 15% ARVC) (26). The diagnostic yield among families depended upon the age of the decedent ranging from a high of 70% when the decedent was between ages 1 and 10 years to a low of 21% when the decedent was between 41 and 49 years of age. Many of these sudden death victims had antecedent warning signs prior to their own death, including syncope in 15% and a family history of young sudden death in 29%. However, there was no prior clinical diagnosis of an inherited cardiac disease for either the decedent or any other family member. In 2014, van der Werf et al., reported a low rate of cardiac events among 405 (267 were <50 years of age) first-degree relatives from 83 of their diagnosis-negative families from their 2010 study (27). Eleven of the 405 (2.7%) relatives died of a cardiac or unknown cause during a median follow-up period of 6.6 years; 5 were over the age of 75 years. Only one individual was younger than 50 years (0.4% [1/267] of this age group), a 10-year-old female who died suddenly while watching television. Her cardiologic evaluation performed 3 months prior was unremarkable. This young girl was identified as belonging to a particularly malignant family where four of seven children have experienced either a SUD or successfully resuscitated cardiac arrest prior to their 17th birthday. In addition, van der Werf reported that sudden death occurred in four non–first-degree relatives in four families with all sudden deaths occurring during the fourth decade of life (27).
In 2012, Caldwell et al. diagnosed 45 (23%) patients from 38 (35%) families with an inheritable cause of sudden death following their cardiologic evaluation of 193 individuals from 108 families because of SUD or aborted cardiac arrest in a young relative (28). Among 146 relatives from 84 families where the index case was found to have a structurally normal heart, 31 (21%) individuals from 25 (30%) families were diagnosed. The most common diagnosis was LQTS (12/31, 39%) followed by HCM (6/31, 19%), CPVT (5/31, 16%), DCM (4/31, 13%), BrS (2/31, 6.5%), ARVC (1/31, 3%), and LV noncompaction (1/31, 3%). Evaluation of the relatives of SUD victims with postmortem evidence of structural heart disease revealed an inherited cause in 14/47 (30%) individuals in 10/24 (42%) families. The most common diagnosis among the 14 individuals was ARVC (6/14, 43%) followed by HCM (5/14, 36%) and DCM (3/14, 21%) (28).
Incomplete penetrance and variable expression are hallmarks of the various cardiac channelopathies which lead to “concealed” forms of these disorders (29). Therefore, clinical assessment of surviving family members of SUD victims may be insufficient to detect LQTS, CPVT, or BrS in unsuspected individuals. A molecular autopsy involving targeted cardiac channel genetic testing is a tool for the forensic pathologist/medical examiner/coroner in order to try to provide the answer to unexplained deaths in the young and subsequently benefit other family members.
Molecular Autopsy Investigations of SUD
In 2004, Chugh et al. identified 12 cases of SUD following a comprehensive postmortem analysis of a consecutive series of 270 adult (age ≥20 years) cases of SCD occurring over a 13-year period (21). Postmortem genetic analysis of the LQTS-susceptibility genes revealed an identical KCNH2 mutation in 2 of these 12 (17%) cases of autopsy-negative SUD. Similarly, Di Paolo et al. performed LQTS molecular autopsies on 10 cases of juvenile (ages 13 to 29 years) SUD and identified KCNQ1 mutations in two individuals (30).
In 2007, Tester and Ackerman completed the largest molecular autopsy series of SUD to date (31,32). Comprehensive mutational analysis of all 60 translated exons in the LQTS-associated genes (KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2) along with targeted analysis of the CPVT1-susceptibility gene (RYR2) was conducted on a series of 49 medical examiner referred cases of SUD. Over one-third of these SUD cases had a presumably pathogenic cardiac channel mutation with mutations in RYR2, alone, accounting for nearly 15% of the cases (31,33). In this series, sudden death was the sentinel event in all but four mutation-positive SUD cases. However, many had a positive family history of cardiac events yet no family members had been diagnosed with either LQTS or CPVT. Overall, approximately half of the 17 decedents, with a cardiac channel mutation detected by postmortem genetic testing, exhibited potential warning signs, either personally or in the family. The repeated observations of unheeded warning signs of syncope and/or family history of sudden death in nearly 15% to 30% of young SUD victims emphasize the importance of heeding potential warning signs.
Population-based studies involving the evaluation of relatives and molecular autopsy investigations of the decedent show that identifiable and potentially treatable cardiac channelopathies (including LQTS, CPVT, and BrS) account for approximately one-third of autopsy-negative SUD in the young. In addition, approximately 10% to 15% of SIDS may be due to these cardiac channelopathies (19,34,35,36,37,38,39,40).
Since an accurate diagnosis from a postmortem molecular analysis of an SUD victim may benefit surviving family members who may also be susceptible to life-threatening arrhythmia syndromes, recent guidelines suggest that postmortem genetic testing should become a standard of care in the evaluation of SUD cases
(24,41,42,43). With now over 100 sudden death-susceptibility genes discovered, next-generation WES, allowing the simultaneous genetic analysis of an individual’s entire library of ∼20,000 genes, is an attractive, cost-effective ($1,000 to $2,000 per sample), and time conducive (few weeks) technique for a comprehensive postmortem genomic study (44). In fact, we recently provided the first ever proof-of-principle case report of a WES-based comprehensive molecular study of a previously healthy 16-year-old sudden death victim, where we identified a pathogenic MYH7 mutation, previously associated with familial HCM, sudden death, and impaired MHC-beta actin-translocating and actin-activated ATPase activity (44). Subsequently, Bagnall et al. completed a WES-based postmortem genetic analysis in their cohort of 28 SUD cases and identified 2/28 (7%) cases with rare variants among the most common LQTS genes (KCNQ1, KCNH2, and SCN5A) and another 6 rare variants among an additional 25 common cardiac channelopathy/cardiomyopathy genes in 6 (21%) SUD cases (45). While the comprehensive nature of WES may be beneficial, it creates a daunting task of scrutinizing thousands of nonsynonymous variants per case. In fact, in our recent exome-based molecular analysis of 14 autopsy-negative SUD cases, each case hosted ∼12,000 nonsynonymous (amino acid altering) variants with an average of 80 such variants localizing to 117 sudden death-susceptibility genes (46). Furthermore, 50% of the SUD cases hosted at least one ultra-rare variant among the 117 sudden death-susceptibility genes with nearly 43% having mutations in cardiomyopathy-associated genes, despite having an autopsy examination void of any overt structural pathology. Therefore, it must be keenly understood that WES findings must be interpreted with extreme caution, particularly in the setting of an autopsy-negative SUD. Without proper interpretation, WES-based postmortem analysis may potentially lead to an inappropriate genetic diagnosis in the SUD case and subsequently in surviving family members who may undergo confirmatory genetic testing, despite being phenotypically normal.
(24,41,42,43). With now over 100 sudden death-susceptibility genes discovered, next-generation WES, allowing the simultaneous genetic analysis of an individual’s entire library of ∼20,000 genes, is an attractive, cost-effective ($1,000 to $2,000 per sample), and time conducive (few weeks) technique for a comprehensive postmortem genomic study (44). In fact, we recently provided the first ever proof-of-principle case report of a WES-based comprehensive molecular study of a previously healthy 16-year-old sudden death victim, where we identified a pathogenic MYH7 mutation, previously associated with familial HCM, sudden death, and impaired MHC-beta actin-translocating and actin-activated ATPase activity (44). Subsequently, Bagnall et al. completed a WES-based postmortem genetic analysis in their cohort of 28 SUD cases and identified 2/28 (7%) cases with rare variants among the most common LQTS genes (KCNQ1, KCNH2, and SCN5A) and another 6 rare variants among an additional 25 common cardiac channelopathy/cardiomyopathy genes in 6 (21%) SUD cases (45). While the comprehensive nature of WES may be beneficial, it creates a daunting task of scrutinizing thousands of nonsynonymous variants per case. In fact, in our recent exome-based molecular analysis of 14 autopsy-negative SUD cases, each case hosted ∼12,000 nonsynonymous (amino acid altering) variants with an average of 80 such variants localizing to 117 sudden death-susceptibility genes (46). Furthermore, 50% of the SUD cases hosted at least one ultra-rare variant among the 117 sudden death-susceptibility genes with nearly 43% having mutations in cardiomyopathy-associated genes, despite having an autopsy examination void of any overt structural pathology. Therefore, it must be keenly understood that WES findings must be interpreted with extreme caution, particularly in the setting of an autopsy-negative SUD. Without proper interpretation, WES-based postmortem analysis may potentially lead to an inappropriate genetic diagnosis in the SUD case and subsequently in surviving family members who may undergo confirmatory genetic testing, despite being phenotypically normal.
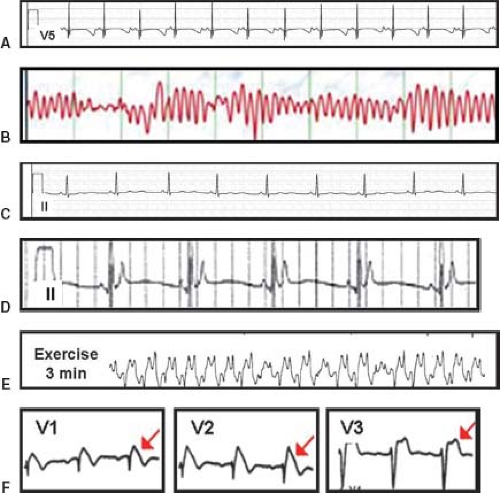 Figure 20.2 Signature electrocardiographic features of various channelopathies. A: QT prolongation—an example of a patient with extreme QT prolongation (QTc >650 ms). Note, the ST segment, T-wave morphology would predict LQT3 but this patient is among the 25% who are genotype negative. Also, the computer-calculated QTc was 362 ms underscoring the mandate to independently compute the QTc. B: Torsade de pointes (“twisting of the points”)—hallmark arrhythmia of LQTS. C: Abnormal U wave in ATS—although subtle, this lead II recording is quite abnormal characterized by normal QT interval, long isoelectric segment between the end of the T wave on the start of the U wave, and long duration U wave in this female with an ATS1-associated mutation in KCNJ2. D: Short QT interval—QTc of approximately 250 ms. E: Exercise-induced bidirectional VT seen in CPVT. F: Coved ST segment elevation in BrS (type I Brugada ECG pattern) in precordial leads V1 and V2 and a saddle-back profile (type II Brugada ECG pattern) in V3. (From Ackerman MJ. Heritable cardiac channelopathies. In: Wyszynski DF, Correa-Villasenor A, Graham TP, eds. Congenital heart defects: from origin to treatment. Oxford; New York: Oxford University Press, 2010:131–140, with permission (50).) |
The Cardiac Channelopathies
The discipline of Cardiac Channelopathies (coined by Clapham and Ackerman) unofficially commenced in 1995 with the discovery of mutations in genes encoding critical ion channels of the heart as the pathogenic basis for congenital LQTS (47,48). Besides classical autosomal dominant (Romano–Ward) LQTS and autosomal recessive (Jervell and Lange-Nielsen) LQTS, the cardiac channelopathies now include Andersen–Tawil syndrome (ATS), Timothy syndrome (TS), drug-induced torsades des pointes (DI-TdP), short QT syndrome (SQTS), CPVT, BrS, idiopathic ventricular fibrillation (IVF), early repolarization syndrome (ERS), progressive cardiac conduction disease (PCCD) or familial atrioventricular conduction block (FAVCB) or Lev-Lenegre disease, and familial atrial fibrillation (FAF). Even primary cardiomyopathies like dilated cardiomyopathy (DCM) can be due to genetically mediated perturbations in ion channels, specifically the SCN5A-encoded cardiac sodium channel (37,38,49).
Overall, cardiac channelopathies may affect as many as 1 in 1,000 persons, may lie dormant for decades or present with SIDS, and may or may not manifest signature electrocardiographic features (Fig. 20.2). Collectively, these cardiac channelopathies represent treatable conditions when recognized. In general, the cardinal events of the cardiac channelopathies comprise syncope, seizures, and SCD. Unfortunately, SCD can be the sentinel event.
Autosomal Dominant Long QT Syndrome (Romano–Ward)
Congenital LQTS is the prototypic cardiac channelopathy with an estimated prevalence of 1 in 2,000 to 2,500 persons. Clinically, LQTS is characterized by abnormal cardiac repolarization resulting in QT interval prolongation (Fig. 20.2A) which predisposes patients to torsade de pointes (TdP, Fig. 20.2B). Palpitations seldom
represent the sole indicator of an episode of TdP. The most common form of LQTS is autosomal dominant LQTS, previously known as Romano–Ward syndrome (51,52).
represent the sole indicator of an episode of TdP. The most common form of LQTS is autosomal dominant LQTS, previously known as Romano–Ward syndrome (51,52).
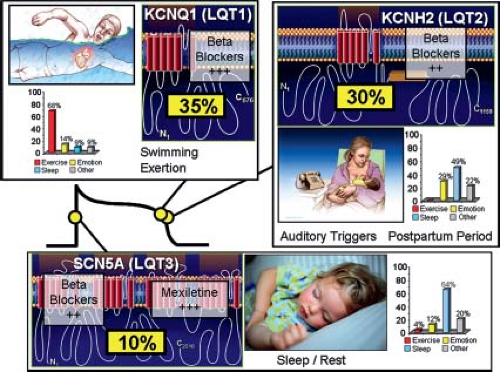 Figure 20.3 Genotype–phenotype correlations in long QT syndrome. Seventy-five percent of clinically strong LQTS is due to mutations in three genes (35% KCNQ1, 30% KCNH2, and 10% in SCN5A) encoding for ion channels that are critically responsible for the orchestration of the cardiac action potential. Genotype–phenotype correlations have been observed, including swimming/exertion and LQT1, auditory triggers/postpartum period and LQT2, and sleep/rest and LQT3. The bar graphs represent genotype–phenotype data from reference 52. Also illustrated is the relative gene-specific effectiveness in beta-blocker therapy where beta-blockers are extremely protective in LQT1 patients and moderately protective in LQT2 and LQT3. Direct late sodium current blockers like mexiletine, flecainide, and ranolazine may be protective in LQT3. (From Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123(9):1021–1037, with permission (70).) |
Hundreds of mutations in 16 distinct LQTS-susceptibility genes have been identified and generally involve either loss-of-function potassium channel mutations or gain-of-function sodium channel mutations. Most recently, gain-of-function mutations in the CACNA1C-encoded L-type calcium channel alpha-subunit have been implicated in pure, “non-Timothy syndrome” LQTS and mutations in three distinct genes (CALM1, CALM2, and CALM3) encoding for the identical calmodulin protein are LQTS-causative (53,54,55). Except for five rare subtypes that stem from perturbations in key cardiac channel interacting proteins (ChIPs) or structural membrane scaffolding proteins (ankyrin B-, calmodulin-, caveolin 3-, yotiao-, and syntrophin alpha-LQTS) (54,55,56,57,58,59), LQTS is a pure “channelopathy” resulting from mutations in cardiac channel alpha and beta subunits. The majority of LQTS is due to mutations in either the KCNQ1-encoded IKs potassium channel (LQT1, 30% to 35%), the KCNH2-encoded IKr potassium channel (LQT2, 25% to 30%), or the SCN5A-encoded INa sodium channel (LQT3, 5% to 10%) (60,61).
The past two decades of LQTS research have provided numerous genotype–phenotype relationships (Fig. 20.3). Genotype–ECG relationships include broad-based T waves in LQT1, low-amplitude or notched T waves in LQT2, and long ST isoelectric segment with normal T-wave morphology in LQT3 (62,63). Gene-specific arrhythmia triggers have been observed including: exertion in LQT1 (particularly swimming), auditory triggers and the postpartum period in LQT2, and events during sleep in LQT3 (64,65,66,67,68). Importantly, the response to beta-blockers is influenced strongly by the underlying genotype where patients with LQT1 realize superior protection than patients with either LQT2 or LQT3 (69).
TABLE 20.1 Summary of Class I Indications for LQTS Genetic Testing (42) | |||
|---|---|---|---|
|
In the presence of a clinical diagnosis of LQTS, the yield of LQTS genetic testing is about 75% (71). Generally accepted indications for LQTS genetic testing are summarized in Table 20.1. However, genetic tests for LQTS and the other cardiac channelopathies must be scrutinized and interpreted with great caution and must not be viewed as binary tests (42,70,72). These genetic tests are probabilistic tests. Some mutations will be definite disease-causative mutations while other genetic variants may not be pathogenic (70). We estimate that at least 10% of the mutations published as pathogenic may lack sufficient evidence to warrant that designation.
Individuals with LQTS may or may not display the hallmark repolarization abnormality of QT interval prolongation (Fig. 20.2A). In fact, approximately 40% to 50% of patients with genotype-positive LQTS have a normal resting QTc (29). This observation reinforces the critical role of genetic testing. In general, a heart rate corrected QT interval (QTc) >480 ms in postpubertal females or >470 ms in postpubertal males should prompt a thorough investigation for LQTS. These values represent the 99th percentile in the distribution of QTc values. Previously, a QTc >440 ms (males) or
>450 ms (females) has been considered “borderline” QT prolongation. In fact, the 2009 AHA/ACC/HRS guidelines denote that a QTc ≥450 ms in adult males and ≥460 ms in adult females be considered “prolonged QTc.” Although 50% of patients with genetically proven LQTS have a QTc <460 ms, these cutoff values would result in an unacceptably high rate of false-positives if used as part of a screening program. Using these cutoff values, an estimated 15% to 20% of the entire population would have “borderline QT prolongation” and 5% to 10% of adults would satisfy the guidelines definition of “prolonged QTc” (Fig. 20.4). In addition, these QTc values are based upon an accurate measurement of the QTc (73). It is absolutely critical to measure the QTc manually. Relying on the computer-derived QTc is unacceptable. Calculation of an average QTc from either lead II or V5 is recommended. Simply using the beat yielding the maximum QTc yields too many false-positives. Furthermore, these QTc distributions do not apply to a maximum QTc value derived from a 24-hour ambulatory ECG recording.
>450 ms (females) has been considered “borderline” QT prolongation. In fact, the 2009 AHA/ACC/HRS guidelines denote that a QTc ≥450 ms in adult males and ≥460 ms in adult females be considered “prolonged QTc.” Although 50% of patients with genetically proven LQTS have a QTc <460 ms, these cutoff values would result in an unacceptably high rate of false-positives if used as part of a screening program. Using these cutoff values, an estimated 15% to 20% of the entire population would have “borderline QT prolongation” and 5% to 10% of adults would satisfy the guidelines definition of “prolonged QTc” (Fig. 20.4). In addition, these QTc values are based upon an accurate measurement of the QTc (73). It is absolutely critical to measure the QTc manually. Relying on the computer-derived QTc is unacceptable. Calculation of an average QTc from either lead II or V5 is recommended. Simply using the beat yielding the maximum QTc yields too many false-positives. Furthermore, these QTc distributions do not apply to a maximum QTc value derived from a 24-hour ambulatory ECG recording.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


