Chapter 2 Basic Aspects of Cellular and Molecular Biology
Molecular Biology
The Genetic Basis of Lung Disease
Genetic factors play an important role in diseases that affect the airways (asthma, chronic obstructive pulmonary disease [COPD], cystic fibrosis, primary ciliary dyskinesia), parenchyma (pulmonary fibrosis, Birt-Hogg-Dubé syndrome, tuberous sclerosis), and vasculature (hereditary hemorrhagic telangiectasia) of the lung (Table 2-1). Such conditions include simple monogenic disorders such as Kartagener syndrome and α1-antitrypsin deficiency, in which mutations of critical genes are sufficient to induce well-defined disease phenotypes. By contrast, many other disease processes affecting the lung are complex genetic traits in which inheritance subtly affects pathogenesis. This group of entities includes COPD, asthma, and idiopathic pulmonary fibrosis. Extending current understanding of the genetic basis of pulmonary conditions will be essential to provide new insights into their underlying pathophysiology, to make predictions about outcome, and to develop novel therapeutic strategies.
Single-Gene Disorders and Respiratory Disease
Many single-gene disorders have been linked with respiratory disease (see Table 2-1). They are perhaps best typified by the autosomal recessive condition α1-antitrypsin deficiency. This condition shows a clear genotype-phenotype correlation with current understanding of the molecular basis providing new insights into the pathogenesis of disease. α1-Antitrypsin is the archetypal member of the serine proteinase inhibitor (“serpin”) superfamily. It is synthesized in the liver and secreted into the plasma, where it is the most abundant circulating proteinase inhibitor. Most people of North European descent carry the normal M allele, but 1 in 25 carries the Z variant (Glu342Lys), which results in plasma α1-antitrypsin levels in the homozygote that are 10% to 15% of the normal M allele. The Z mutation causes the accumulation of α1-antitrypsin in the rough endoplasmic reticulum of the liver, predisposing the homozygote to the development of juvenile hepatitis, cirrhosis, and hepatocellular carcinoma. The greatly reduced circulating levels of α1-antitrypsin are unable to protect the lungs against proteolytic damage by neutrophil elastase, predisposing the Z homozygote to the development of early-onset emphysema.
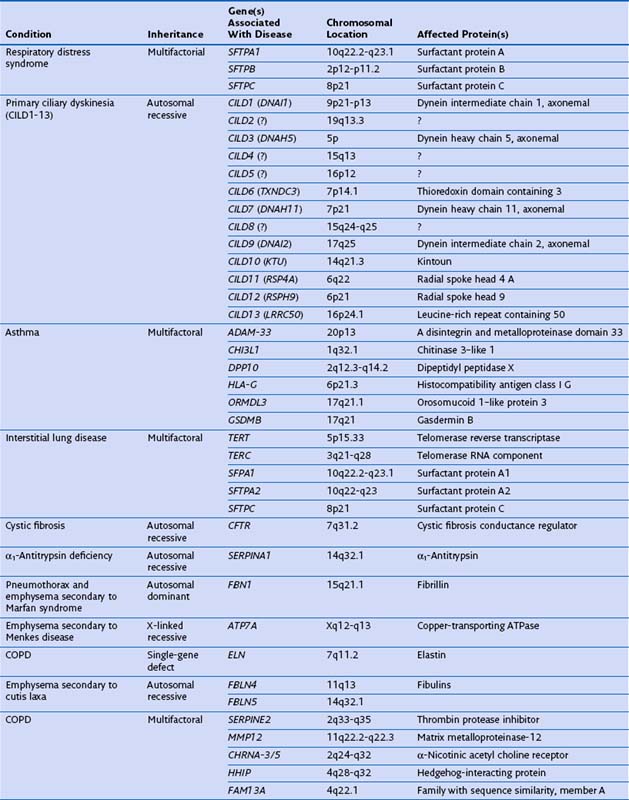
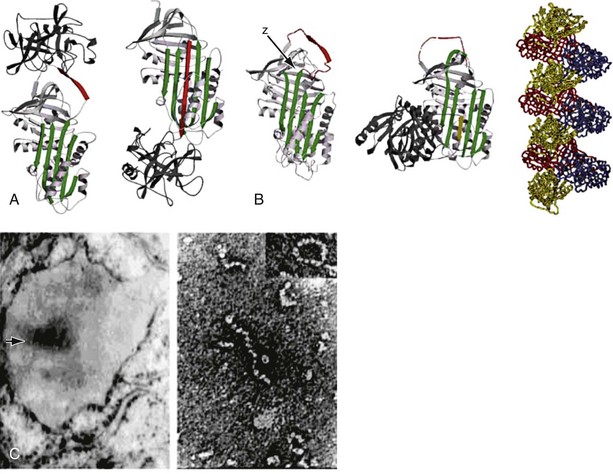
The structure of α1-antitrypsin is based on a dominant β-pleated sheet A and nine α-helices (Figure 2-1). This scaffold supports an exposed mobile reactive loop that presents a peptide sequence as a pseudosubstrate for the target proteinase. After docking, the proteinase is inactivated by a mousetrap-type action that swings it from the top to the bottom of the serpin in association with the insertion of an extra strand into β-sheet A (see Figure 2-1). This six-stranded protein bound to its target enzyme is then recognized by hepatic receptors and cleared from the circulation. The structure of α1-antitrypsin is central to its role as an effective antiproteinase but also renders it liable to undergo conformational change in association with disease. The Z mutation is at residue P17 (17 residues proximal to the key P1 amino acid that defines the inhibitory specificity of α1-antitrypsin) at the head of a strand of β-sheet A and the base of the mobile reactive loop (see Figure 2-1). The mutation opens β-sheet A, thereby favoring the insertion of the reactive loop of a second α1-antitrypsin molecule to form a dimer (see Figure 2-1). This dimer can then extend to form polymers that tangle in the endoplasmic reticulum of the liver to form the inclusion bodies resulting in liver disease. Support for this pathomechanism comes from the demonstration that Z α1-antitrypsin formed chains of polymers when incubated under physiologic conditions. The rate was accelerated by raising the temperature to 41° C and could be blocked by peptides that compete with the loop for annealing to β-sheet A. The role of polymerization in vivo was clarified by the finding of α1-antitrypsin polymers in inclusion bodies from the livers of Z α1-antitrypsin homozygotes (see Figure 2-1).
Gene Hunting for Complex Genetic Diseases and Its Pitfalls: COPD and Asthma
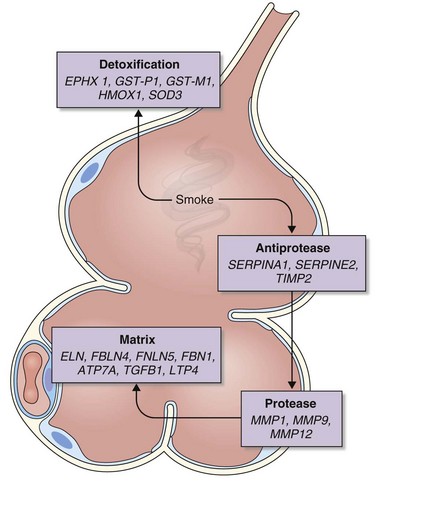
The analysis was made more complex in COPD by the inherent complexity of the disease phenotype—a heterogeneous mix of airway disease and emphysema. Indeed, larger family-based studies have shown the independent clustering of the airway disease and emphysema components of COPD within families. This finding suggests that different genetic factors predispose to each of these components of the phenotype. The only way to overcome the inherent variation in COPD is to focus on groups of patients with well-characterized disease components or to undertake studies with large sample sizes and then to replicate any positive findings in other cohorts. This is now the case with candidate gene studies, and good evidence has emerged to show that heterozygosity for α1-antitrypsin deficiency (phenotype PiMZ) and polymorphisms in genes involved in oxidative stress—those encoding microsomal epoxide hydrolase (EPHX1), glutathione S-transferase (GST-P1 and GST-M1), heme oxygenase (HMOX1), and superoxide dismutase 3 (SOD3)—are associated with an increased risk of COPD (Figure 2-2). More recently, a minor allele of an SNP in the matrix metalloprotease-12 gene (MMP12) has been shown to protect against COPD in adult smokers.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


