13 Bare Metal and Drug-Eluting Coronary Stents
 Abbreviations
Abbreviations Introduction
Introduction Overview of Bare Metal Stents
Overview of Bare Metal Stents
Limitations of Balloon Angioplasty and Development of the Coronary Stent
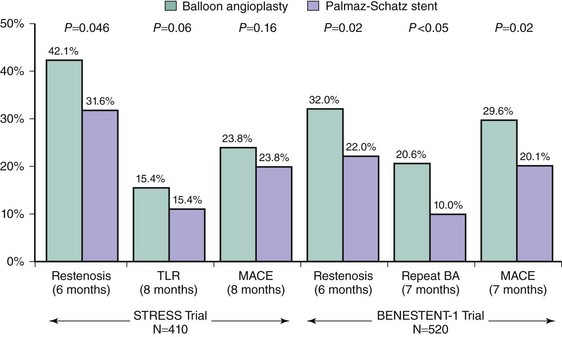
The performance of the first successful balloon angioplasty by Andreas Grüntzig on September 16, 1977, in Zurich, Switzerland, was a landmark event heralding the inception of percutaneous management of obstructive coronary artery disease. Although this initial procedure set the stage for the millions of PCI procedures that have since taken place, stand-alone balloon angioplasty as performed by Grüntzig and other early pioneers was a highly unpredictable experience. While the majority of vessels tolerate the focal plaque dissections caused by balloon dilatation and heal sufficiently to result in an adequate lumen, the injury to the vessel wall can be excessive and, along with acute recoil and chronic constrictive remodeling, can result in balloon angioplasty’s two major limitations: abrupt closure (occurring acutely or within the first several days after angioplasty) and restenosis (occurring later, within months after the procedure). The coronary stent was thus devised as an endoluminal scaffold to create a larger initial lumen, seal dissections, and resist recoil and late vascular remodeling, thereby improving upon the early and late results of balloon angioplasty. The first implantation of stents in human coronary arteries occurred in 1986 when Ulrich Sigwart, Jacques Puel, and their colleagues placed the Wallstent sheathed self-expanding metallic mesh scaffold (Medinvent, Lausanne, Switzerland) in the peripheral and coronary arteries of eight patients.1 Further experience with this device demonstrated high rates of thrombotic occlusion and late mortality,2 although patients without thrombosis had a 6-month angiographic restenosis rate of only 14%. This suggested for the first time that stenting could improve late patency in addition to stabilizing the acute results obtained after conventional balloon angioplasty. Contemporaneously, Cesare Gianturco and Gary Roubin developed a balloon-expandable coil stent consisting of a wrapped stainless steel wire resembling a clamshell. Completion of a phase II study using the Gianturco-Roubin stent to reverse postangioplasty acute or threatened vessel closure3 led to U.S. Food and Drug Administration (FDA) approval for this indication in June 1993. While these stents were being developed and tested, Julio Palmaz designed a balloon-expandable stainless steel stent in 1984 that became the first “slotted tube stent.” In this design, rectangular slots were cut into thin-walled stainless steel tubing such that balloon inflation within the stent deformed the rectangular slots into diamond-shaped windows or cells. Although this design allowed for relatively straightforward deployment, the rigidity of the stent made it difficult to deliver it to the coronary vasculature. In 1987 Richard Schatz modified this design by placing a 1-mm central articulating bridge connecting the two rigid 7-mm slotted segments,4 thus creating the 15-mm Palmaz-Schatz stent (Johnson and Johnson Interventional Systems, Warren, NJ) (Fig. 13-1). The first coronary Palmaz-Schatz stent was placed in a patient by Eduardo Sousa in São Paulo, Brazil, in 1987. The Palmaz-Schatz stent was subsequently investigated in two landmark randomized trials comparing balloon angioplasty to elective stenting. In the STRESS and BENESTENT-1 studies, the use of the Palmaz-Schatz stent was associated with a 20% to 30% reduction in clinical and angiographic restenosis compared with conventional balloon angioplasty (Fig. 13-2).5,6 The Palmaz-Schatz stent also resulted in markedly improved initial angiographic results, with a larger postprocedural minimal luminal diameter and fewer residual dissections, which translated into a lower rate of subacute vessel closure. These results led to approval of the Palmaz-Schatz Stent by the FDA in 1994. Long-term follow-up (through 15 years) has subsequently demonstrated few late clinical or angiographic recurrences from years 1 to 5 after coronary stent implantation,7,8 with slight and progressive decrements in luminal size thereafter extending beyond 10 years.9 The mechanisms of this late progression of disease are not entirely known, but as overall stent thrombosis rates remained low in these patients (1.5% at 15 years), the development of new atherosclerosis within the stented segment as a cause of late restenosis has been hypothesized.9 Despite the success of the Palmaz-Schatz stent in improving the early and late results of conventional balloon angioplasty, widespread adoption of stent technology was initially hindered by high rates of subacute stent thrombosis, necessitating an intense antithrombotic and antiplatelet regimen (consisting of aspirin, dextran, dipyridamole, heparin, and warfarin). Further refinements in the stent procedure and periprocedural pharmacotherapy regimens were thus required. Antonio Colombo and colleagues demonstrated reduced rates of stent thrombosis with more aggressive intravascular ultrasound (IVUS)-guided deployment techniques, including routine high-pressure adjunctive dilatation (>14 atmospheres),10 along with the use of aspirin and a second antiplatelet agent (the thienopyridine ticlopidine) rather than prolonged warfarin therapy. These modifications significantly reduced the incidence of stent thrombosis to about 1% to 2%, along with a marked reduction in bleeding and femoral arterial complications.11 The confirmation of these initial findings in several randomized clinical trials definitively established the superiority of dual antiplatelet therapy (with aspirin and ticlopidine) over anticoagulation with warfarin for the prevention of stent thrombosis and facilitated the widespread adoption of this therapy for coronary stenting by the late 1990s.12–15

Stent Composition
Until quite recently, the most widely used stent material was 316L stainless steel. Now, however, cobalt chromium and platinum chromium alloys have been employed to provide for lower-profile thin stent struts (75-µm versus 100- to 150-µm in most stainless steel stents) that maintain strength and visibility. Most self-expanding stents utilize nitinol, a nickel-titanium alloy, which, after being baked at a high temperature, maintains shape memory for a preset size and configuration. Other than gold (which has been shown to increase restenosis), there is little evidence that thrombosis or restenosis rates vary with the specific stent metal, although the final stages of surface finishing, smoothing, and purification or passivation may affect early thrombotic and late restenotic processes.16 There has also been increased interest in polymeric biodegradable stents, which theoretically offer the advantages of increased longitudinal flexibility (though at the expense of radial force), compatibility with noninvasive imaging, and complete bioabsorption over a period of months to a year or longer, thereby restoring underlying vascular reactivity (see Chap. 33, “Biodegradable Stents.”)
Stent Configuration and Design
Stents can be assigned to one of three subgroups, based on construction: wire coils, slotted tubes/multicellular, and modular designs. The vast majority of stents in current use are either slotted tube/multicellular or modular in design. Slotted tube/multicellular stents are often classified as having open or closed cells. Open-cell designs tend to have varying cell sizes and shapes, which provide increased flexibility, deliverability, and side branch access, with staggered cross-linking elements to provide radial strength. Closed-cell designs typically incorporate a repeating unicellular element that provides more uniform wall coverage with less tendency for plaque prolapse, but at the expense of flexibility and side branch access. Stent design may significantly impact acute and late vascular responses. Stents that possess better conformability, less rigidity, and greater circularity experimentally produce less vascular injury, thrombosis, and neointimal hyperplasia.17,18 Clinical studies have suggested that thin stent struts may be associated with reduced neointimal hyperplasia and lower rates of restenosis.19
Stent Coatings
A variety of coatings have been used to attempt to reduce the thrombogenicity or restenosis of metallic stents (Table 13-1). Experimental studies have demonstrated that the coating of stents with inert polymers may reduce surface reactivity and thrombosis,18 although until recently, most polymers used were found to provoke intense inflammatory reactions.20 With the advent of the DES came a renewed interest in the study of stent coatings, primarily to act as drug-carrier vehicles. However, concerns regarding the long-term safety of drug-eluting stents and the requirement for extended-duration dual antiplatelet therapy have led to a renewed interest in biocompatible stent coatings. A number of novel stent coatings are currently under investigation. Additionally, covered stents (metallic stents covered by a distensible microporous polytetrafluoroethylene [PTFE] membrane) are of unquestioned clinical utility in treating life-threatening perforations. They are also used for excluding giant aneurysms, pseudoaneurysms, or clinically significant fistulas (see Chap. 27, “Complications of Coronary Intervention.”)
TABLE 13-1 Stent Coatings Designed to Reduce Thrombogenicity
Balloon-Expandable versus Self-Expanding Stents
Balloon-expandable stents are mounted onto a delivery balloon and delivered into the coronary artery in their collapsed state. Once the stent is in the desired location, inflation of the delivery balloon expands the stent and embeds it in the arterial wall, following which the stent delivery system is removed. Almost all stents implanted in human coronary arteries are balloon-expandable. Self-expanding stents incorporate either specific geometric designs or nitinol shape-retaining metal to achieve a preset diameter and are released from a restraining sheath once placed in position. Although self-expanding stents are flexible and often easier to deliver compared with their balloon-expandable counterparts, restenosis has remained a concern, thus limiting their use in coronary arteries.21 Recently, however, a renewed interest in self-expanding stents with reduced outward expansion force for the treatment of patients with acute coronary syndromes or vulnerable plaque has surfaced.22
 Indications for Coronary Stenting and Comparison with Balloon Angioplasty
Indications for Coronary Stenting and Comparison with Balloon Angioplasty
Stenting after Failed Balloon Angioplasty
Stents may be used either on a routine (planned) basis or after failed balloon angioplasty for acute or threatened vessel closure (“bail-out” stenting). One of the major benefits of stenting is the ability to reverse abrupt closure due to dissection and recoil, thus eliminating the need for high-risk emergency bypass surgery.23 These data—coupled with the fact that routine stent implantation compared with balloon angioplasty provides superior acute results and greater event-free survival in almost every patient and lesion subtype studied to date—has for the most part relegated balloon dilation to the rare lesion that is too small (<2.25 mm) for stenting, or to which a stent cannot be delivered because of excessive vessel tortuosity or calcification, or in patients in whom thienopyridines are contraindicated.
Routine Stenting during PCI
The utility of routine stent implantation as a modality to reduce acute vessel closure and late restenosis was first demonstrated in the STRESS and BENESTENT-1 trials, which enrolled patients undergoing PCI of discrete focal lesions.5,6 As a result, the types of lesions treated in these trials (discrete de novo lesions coverable by one stent, with reference vessel diameter [RVD] 3.0–4.0 mm) became known as “Stress/Benestent” lesions to differentiate them from more complex stenoses. Despite initial concerns regarding the diminished efficacy of coronary stents (which were more costly than balloon angioplasty alone) with more generalized use of these devices,24 abundant randomized and nonrandomized data now exist comparing stenting to balloon angioplasty across a range of patient and lesion subsets, and they almost universally demonstrate an advantage to coronary stenting over conventional balloon angioplasty.25–27
 Overview of Drug-Eluting Stents
Overview of Drug-Eluting Stents
Limitations of Bare Metal Stents
By the late 1990s, for most patients with coronary artery disease, stent implantation became the prevailing treatment because of their more predictable acute and late angiographic results compared with conventional balloon angioplasty, atherectomy, and laser. Improvements in procedural technique, more effective antiplatelet regimens, and the introduction of increasingly lower-profile, more flexible and deliverable devices additionally contributed to the ascendancy of stenting. With improvements in stent deliverability and reductions in stent thrombosis to <1%, restenosis emerged as the major persistent limitation of coronary stenting. Although coronary stents increase acute luminal diameters to a greater extent than balloon angioplasty (greater acute gain), the vascular injury caused by stent implantation elicits an exaggerated degree of neointimal hyperplasia, resulting in greater late decreases in luminal diameter (late loss) compared with balloon angioplasty alone.5,6 Importantly, however, in comparing stents with balloon angioplasty, the mean incremental gain in luminal dimensions with stenting is statistically greater than the mean incremental increase in late loss, resulting in a larger net gain in minimal luminal dimensions. This observation led Kuntz and Baim to formulate the “bigger is better” concept: the greater the acute gain, the lower the ultimate rate of restenosis.28,29 Nonetheless, even with optimal stent implantation, restenosis after BMS implantation still occurred in approximately 20% to 40% of patients within 6 to 12 months, in part because of more complex patient and lesion subsets that were treated with stenting than with balloon angioplasty. Therefore coronary restenosis became known as the Achilles’ heel of coronary stenting, with significant resources devoted to its prevention and treatment (see chapter on restenosis).
DES, which maintain the mechanical advantages of BMS while delivering an antirestenotic pharmacological therapy locally to the arterial wall, have been shown to effectively and safely reduce the amount of in-stent tissue that accumulates after stent implantation, resulting in significantly reduced rates of clinical and angiographic restenosis. These devices were designed specifically to prevent the neointimal hyperplasia resulting after conventional BMS placement and have been highly successful in this regard. In numerous randomized trials, the reduction in neointimal hyperplasia that occurs with DES compared with BMS placement has been shown to result in a marked reduction in binary angiographic restenosis and target lesion revascularization (TLR).30–32 The initial results of the pivotal randomized trials that led to device approval have been replicated and validated in numerous subsequent trials and real-world registries across the spectrum of disease and lesion subtypes.33,34
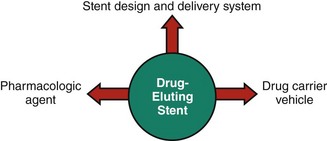
Components of Drug-Eluting Stents
The three critical components of a DES that must be optimized to ensure its safety and efficacy are as follows: (1) the stent itself (including its delivery system), (2) the pharmacologic agent being delivered, and (3) the drug carrier vehicle, which controls the drug dose and pharmacokinetic release rate (Fig. 13-3).
Designs of Drug-Eluting Stents
The stent component of the DES has typically consisted of a predicate BMS without specific modifications. Indeed, first-generation DES designs often appropriated “off-the shelf” stent designs in order to expedite device development and regulatory approval. Subsequent DES have incorporated newer, more flexible designs, with resultant improvements in device delivery and performance.35,36 Additionally, newer dedicated DES designs have included modifications aimed at either optimizing local drug delivery while reducing total drug dose (e.g., abluminal wells engineered into the stent struts) or modifying the stent surface to facilitate direct drug delivery and/or arterial healing following implantation (without a drug carrier vehicle per se).
Basic Pharmacology of Drug-Eluting Stents
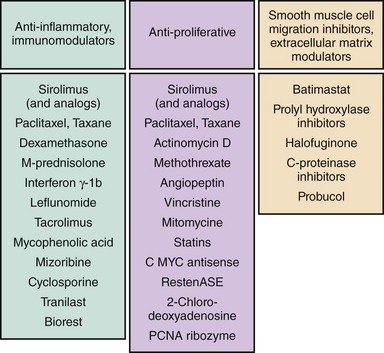
Following promising cell culture and in vitro development, the antirestenotic properties of a wide range of pharmacological agents have been tested in humans (Fig. 13-4). Among these, the two most clinically effective classes of agents are the rapamycin-analogue family of drugs and paclitaxel. The principal mechanism of action of rapamycin (also known as sirolimus) and its analogues (including zotarolimus, everolimus, biolimus A9, and novolimus) is inhibition of the mammalian target of rapamycin (mTOR), which prevents cell cycle progression from the G1 to S phase.37 Two other rapamycin analogues that have been used on DES platforms—tacrolimus and pimecrolimus—have a different mechanism of action, binding directly to FKBP506 and thereby inhibiting the calcineurin receptor with downregulation of cytokines and inhibition of smooth muscle cell activity;38 unlike the mTOR inhibitors, these agents have not shown significant antirestenotic effects. The other agent that has been used effectively not only in DES, but also on drug-eluting balloons is paclitaxel. By interfering with microtubule function, paclitaxel has multifunctional antiproliferative and anti-inflammatory properties; it prevents smooth muscle migration, blocks cytokine and growth factor release and activity, interferes with secretory processes, is anti-angiogenic, and impacts signal transduction.39–41 At low doses (similar to those in DES applications), paclitaxel affects the G0-G1 and G1-S phases (G1 arrest), resulting in cytostasis without cell death.39,42
Drug-Eluting Stent Polymers and Drug Carrier Systems
The inability to predictably deliver a specific dose of active drug to the arterial wall over the right time frame led to the failure of early DES programs.43 To ensure accurate drug dosing, a drug delivery vehicle became necessary, which for most first-generation stents was a durable (nonerodable) polymer. A wide range of polymer systems have since been developed and are DES-specific (as discussed in subsequent sections of this chapter). While the polymer is instrumental in regulating the pharmacokinetics of drug delivery to the arterial wall (which is necessary for reduced neointimal hyperplasia), the polymer may elicit deleterious vascular responses. Specifically, histopathological studies have demonstrated hypersensitivity and eosinophilic inflammatory reactions as well as delayed endothelialization with DES that were not previously seen with BMS.44–46 Whether these maladaptive vascular responses are directly related to the polymer or to toxic reactions from the drug itself is speculative, but in animal models these effects can be attenuated by modification of the polymer vehicle.47 It is believed that in selected patients, excessive inflammation and delayed endothelialization play a role in the development of late stent malapposition, aneurysm formation, and stent thrombosis/restenosis.44,48 For these reasons, there has been great interest in developing inert and biocompatible polymers, bioabsorbable/biodegradable polymers, and even polymer-free DES.
Generational Classification of Drug-Eluting Stents
Given the rapid evolution of DES technologies, DES are often classified into several generations of development (Table 13-2). First-generation devices include the two DES that were initially approved for clinical use by most regulatory bodies, each of which utilized an early BMS stent platform (suboptimal by today’s standards) with a durable polymer (prone to inflammation) to deliver either sirolimus or paclitaxel. Second-generation devices (currently used in the majority of DES procedures) have incorporated more deliverable, thinner-strut stents with polymers designed for biological compatibility. Most second-generation DES utilize rapamycin-analogues, although, as discussed below, there may still be a selective role for paclitaxel-eluting stents (PES). Future-generation DES will continue to undergo iteration, with further modifications of the base stent and use of biodegradable/bioabsorbable or polymer-free drug delivery vehicles.

 First-Generation Drug-Eluting Stents
First-Generation Drug-Eluting Stents
The Cypher Sirolimus-Eluting Stent
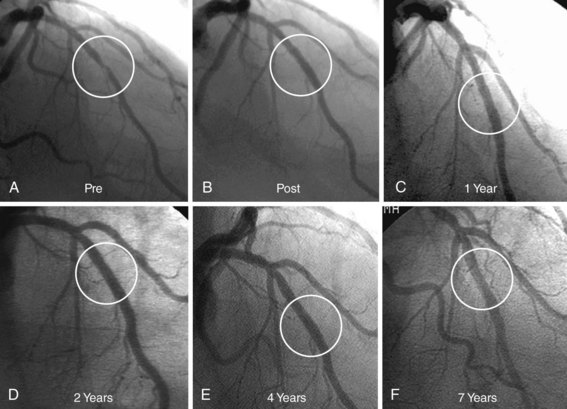
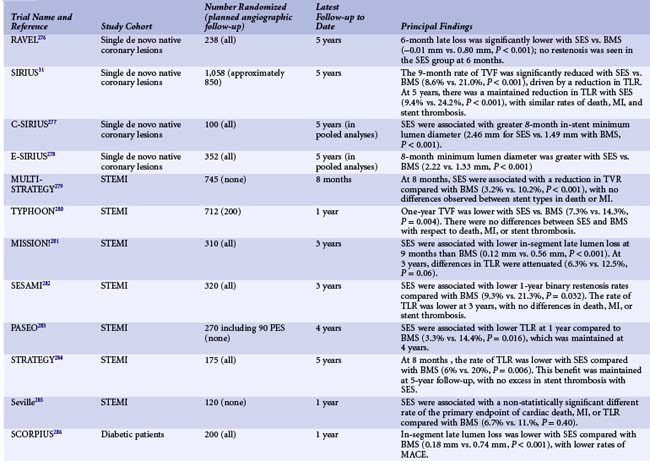
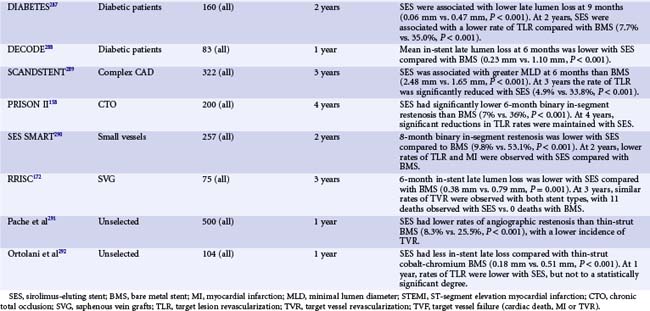
The first important DES was the Cypher stent (Cordis, Johnson and Johnson), with initial first-in-human studies as well as subsequent clinical trials leading to its approval in Europe in 2002 and the United States in 2003. Sirolimus is a highly lipophilic, naturally occurring macrocyclic lactone, which was first isolated from Streptomyces hygroscopicus, found in a soil sample from Easter Island, and was initially developed as an antifungal agent. Shortly thereafter it was discovered that sirolimus also possessed potent immunosuppressive properties. It was initially approved by the FDA as Rapamune for the prevention of renal transplant rejection in 1999. The primary mechanism of action of sirolimus’s inhibition of neointimal hyperplasia is thought to be related to its ability to bind to FK-binding protein-12 (FKBP-12) in cells; the sirolimus-FKBP-12 complex then binds to and inhibits activation of mTOR, preventing progression in the cell cycle from the late G1 to S phase.37 The sirolimus-eluting stent (SES) was demonstrated to have a marked effect on the suppression of neointimal hyperplasia with low toxicity following implantation in initial small and large animal studies.49,50 The base stent platform for the Cypher SES is the Bx-Velocity stent, a slotted tube with a closed cell design constructed from 316L stainless steel. Prior to mounting, the stent is coated on its luminal as well as its abluminal surface (i.e., conformal coating) with biostable (nonerodible) polymers consisting of poly-n-butyl methacrylate and polyethylene-vinyl acetate, which are loaded with 140 mcg/cm2 sirolimus. The polymers, which uniformly coat the stent to a thickness of 5 to 10 microns, serve as a drug carrier vehicle, ensuring that the sirolimus is released into the vessel wall in a programmed manner. The slow-release formulation of the Cypher SES employed in clinical practice uses a base coat of blended polymers, which is loaded with sirolimus, as well as a top coat of polymer alone (without sirolimus), which acts as a diffusion barrier and thus reduces the rate of drug release from the base coat into the vessel wall. With this formulation of polymers, most of the sirolimus (approximately 80%) is released within the first month after stent implantation. Human experience with the CypherSES was first reported from the first-in-man (FIM) study initiated in 1999 in 45 patients with symptomatic de novo lesions < 18 mm in length with an RVD of 3 to 3.5 mm in native coronary arteries at the Institute Dante Pazzanese of Cardiology in São Paulo, Brazil, and the Thoraxcenter, Rotterdam, The Netherlands. In this study, the SES demonstrated marked suppression of neointimal hyperplasia measured by IVUS and quantitative coronary angiography at 4 months and 1, 2, and 4 years.51 Serial angiography and IVUS have now been performed at 7 years, showing continued vessel patency without further late loss (Fig. 13-5). Following the success of SES in this initial study, the landmark RAVEL trial was conducted, in which 238 patients outside the United States with relatively simple de novo coronary lesions (lesion length ≤ 18 mm in coronary arteries 2.5–3.5 mm in diameter) were randomized to either the Cypher SES or the uncoated Bx-Velocity stent.30 In RAVEL, mean late loss at 6 months was markedly lower among SES-treated than BMS-treated patients (-0.01 mm vs. 0.80 mm, P < 0.001), with a corresponding reduction in the rate of restenosis (0% vs. 26%, P < 0.001). The pivotal U.S. randomized trial that led to FDA approval of SES was the SIRIUS trial, a 1,058-patient randomized trial comparing the Cypher SES with the uncoated Bx-Velocity in patients with vessel diameters of 2.5 to 3.5 mm and lesion lengths of 15 to 30 mm.52 The primary endpoint, the rate of target vessel failure (TVF: a composite of cardiac death, myocardial infarction [MI], or target vessel revascularization [TVR]) at 9 months, was markedly lower among SES-treated patients (8.6% vs. 21.0%, P < 0.001). The rate of major adverse cardiac events (MACE) was similarly lower with SES compared with BMS (7.1% vs. 18.9%, P < 0.001). A 60% to 80% relative reduction in composite adverse events with SES versus BMS was observed in all examined subgroups in the trial, including diabetic patients, and did not depend on vessel size or lesion length. Among 703 patients in whom 8-month routine angiographic follow-up was performed, mean in-stent late loss was markedly lower with SES compared with BMS (0.17 mm vs. 1.00 mm, P < 0.001), and in-segment angiographic restenosis was greatly reduced with SES (17.6% vs. 50.5%, P < 0.001). By IVUS, the in-stent percent volumetric obstruction at 8 months was reduced from 33.4% with the Bx Velocity to 3.1% with the SES (P < 0.001). Late stent malapposition was present in 9.7% of Cypher SES patients versus 0% of Bx Velocity patients (P = 0.02). Among patients experiencing restenosis events in SIRIUS, the pattern of restenosis was diffuse in only 13% of patients with SES restenosis versus 58% with BMS restenosis (P < 0.001). Thus treatment with repeat PCI for SES restenosis (typically focal) patients was associated with more favorable outcomes than for BMS-restenosis patients (typically diffuse).53 On the basis of these results, in April 2003 the Cypher SES became the first DES approved by the FDA. To date, this stent has been one of the most studied devices in modern history, with at least 20 randomized trials performed comparing the commercialized Cypher SES to the BMS across a range of patient indications and lesion subsets (Table 13-3). Collectively, these trials demonstrate that the Cypher SES results in a near abolition of in-stent late loss (averaging ~0.15 mm across studies), with an approximate 50% to 75% reduction in angiographic restenosis and clinical recurrence (TLR) compared with bare metal comparators. Longer-term follow-up with this device has extended to 5 years and beyond, particularly for the four major SES trials (RAVEL, SIRIUS, C-SIRIUS, and E-SIRIUS). In these analyses, treatment with SES has resulted in sustained reductions in clinical restenosis endpoints with similar rates of death, MI, and stent thrombosis found in both SES and BMS arms.54
The Taxus Paclitaxel-Eluting Stent
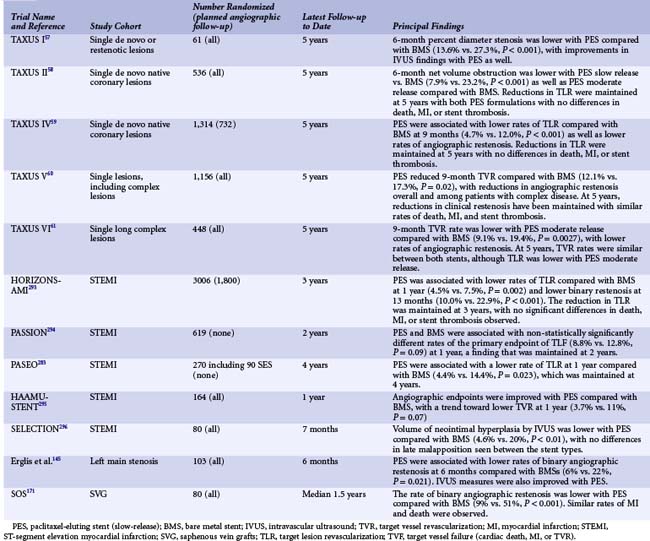
Paclitaxel, a highly lipophilic diterpenoid compound, was first isolated in 1963 from the Pacific yew tree (Taxus brevifolia) and subsequently developed for its potent antineoplastic properties. Paclitaxel is insoluble in water and has thus been combined with an intravenous oil-based cremophor for intravenous injection as the oncological compound Taxol. The principal action of paclitaxel is to interfere with microtubule dynamics, preventing depolymerization. Because microtubules are ubiquitous, paclitaxel has widespread multicellular and multifunctional activities with dose-dependent effects. Paclitaxel has antiproliferative and anti-inflammatory properties, prevents smooth muscle migration, blocks cytokine and growth factor release and activity, interferes with secretory processes, is antiangiogenic, and impacts signal transduction.39–41 At low doses (similar to those in DES applications), paclitaxel affects the G0-G1 and G1-S phases (G1 arrest), resulting in cytostasis without cell death (probably via the induction of p53/p21 tumor suppression genes).39,42 Systemic paclitaxel was shown to inhibit restenosis in a rat carotid injury model at levels more than 100-fold lower than those required for tumor cytotoxicity.40 Neointimal area was greatly reduced in a rabbit balloon injury experiment using local paclitaxel administration,39 and stent-based paclitaxel elution from polymer-based systems has been shown to profoundly reduce intimal hyperplasia in rabbit iliac arteries for up to 6 months with dose-dependent efficacy and toxicity.55,56 The TAXUS paclitaxel-eluting stent (PES) (Boston Scientific, Natick, MA) consists of paclitaxel contained within a polyolefin-derivative biostable polymer (styrene-isobutylene-styrene, referred to as SIBS [Translute]), originally coated on the Nir stent and subsequently on the Express open-cell slotted-tube stainless steel stent platform (the device from which most of the randomized clinical trial data for this stent were derived). The base BMS for the current TAXUS stent is the newer Liberté stent, a more flexible, thinner-strutted closed-cell stainless steel slotted-tube stent. Depending on the relative ratio of paclitaxel to polymer, the stent may be formulated with varying release kinetics. The clinically available formulation of the TAXUS PES is the slow-release (SR) formulation, although the moderate-release (MR) formulation has also been tested in moderate-sized clinical trials. The SR stent has relatively more polymer to drug (paclitaxel concentration of 1 mcg/mm2), with a coat thickness 18 µm and approximately 8% in vivo paclitaxel elution in 30 days. The drug is eluted in a rapid burst phase over the initial 48 hours, followed by a slow, sustained release for the next 10 to 30 days, with the remainder sequestered in the bulk of the polymer matrix below the surface without pathways to the external environment (thus permanently retained on the stent). In a series of porcine experiments at 30, 90, 180, and 360 days involving a total of 350 swine and 800 stents, both SR and MR TAXUS stent formulations were shown to be vasculocompatible, with early development of a thin, mature neointima with low levels of inflammation, microthrombi, and peristent amorphous material deposition (fibrin), no evidence of cytotoxicity, and complete healing and endothelialization within 90 days (data on file, Boston Scientific). The clinical safety and efficacy of the TAXUS PES has been tested in randomized trials in the TAXUS clinical program.57–61 TAXUS I and II evaluated the TAXUS PES on the Nir stent platform in focal lesions, whereas TAXUS IV, V, and VI investigated the TAXUS Express stent in more complex lesions with longer-term follow-up. All studies have used the SR formulation except for one arm of the TAXUS II trial and TAXUS VI. Collectively, these trials demonstrate a marked decrease of binary restenosis with PES compared with BMS, with a ~60% to 75% reduction in the need for TLR. In the pivotal prospective, randomized, double-blind TAXUS IV study, 1,314 patients with single de novo lesions with visually estimated lengths of 10 to 28 mm in native coronary arteries with an RVD of 2.5 to 3.75 mm were assigned to either a TAXUS SR stent or Express BMS control.59 At 9 months, the primary endpoint of target vessel revascularization (TVR) was reduced with the TAXUS PES from 12.0% to 4.7% (P < 0.001). Follow-up angiography at 9 months demonstrated marked reductions in mean in-stent late loss (0.39 vs. 0.92 mm, P < 0.001) and binary in-segment restenosis (7.9% vs. 26.6%, P < 0.001). Restenotic lesions were typically focal in nature with PES (mean length 9.8 mm vs. 15.3 mm with BMS, P < 0.001). Late coronary aneurysms were infrequent in both groups and not increased with PES. By IVUS, the in-stent percent volumetric obstruction at 8 months was reduced from 29.4% with the BMS to 12.2% with the PES (P < 0.001). Late stent malapposition at 9 months was present in 1.1% of PES patients versus 2.2% of BMS patients (P = 0.62). Clinical and angiographic efficacy was present across a broad range of patient and lesion subtypes. On the basis of these results, in March 2004 the TAXUS SR stent became the second DES approved by the FDA. Since device approval, the TAXUS PES has been studied in at least 12 randomized trials comparing this device to the BMS across a range of patient indications and lesion subsets (Table 13-4). These studies have demonstrated consistent reductions in measures of neointimal hyperplasia (measured either by angiography or by IVUS), with resultant reductions in clinical restenosis endpoints compared with the BMS. Longer-term follow-up with this device has extended to 5 years and beyond, particularly for the four major PES trials of the SR stent (TAXUS I, II, IV, and V). In these analyses, treatment with PES results in sustained reductions in clinical restenosis endpoints, with similar rates of death, MI, and stent thrombosis found in both the PES and BMS arms.59 The currently available PES device is the TAXUS Liberté stent (using the same drug and polymer formulation as the TAXUS Express SR but with an improved stent platform). This device was approved for clinical use based on the TAXUS ATLAS program, in which nonrandomized data from several TAXUS Liberté PES single-arm studies were compared with the treatment arms from prior TAXUS trials with the Express PES.62 In routine “workhorse” lesions, the Liberté PES was shown to have similar rates of restenosis and safety endpoints compared with the Express PES. However, when used to treat small vessels and long lesions, the Liberté PES was associated with lower rates of TLR compared with a matched sample of Express PES patients. Additionally, in long lesions, there was a lower rate of MI among patients treated with the Liberté PES at 1 year (1.4% vs. 6.5%, P = 0.02), a finding that has been maintained up to 3 years.63
Comparisons of First-Generation Drug-Eluting Stents and Metanalyses
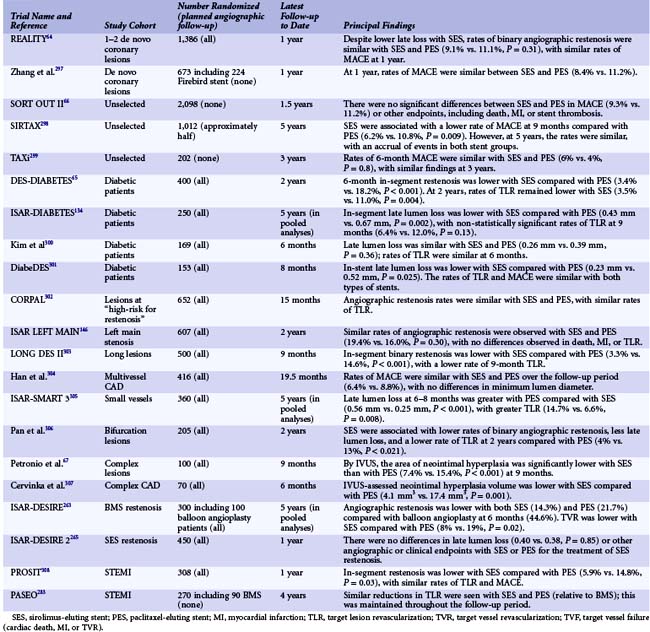
Following the approval of the TAXUS PES for commercial use, a series of comparisons between the two approved devices (Cypher SES and TAXUS PES) ensued in order to determine whether superiority could be established for a particular DES. A total of 21 randomized comparisons of different sizes have been performed to date in numerous patient and lesion cohorts; they have included a variety of IVUS, angiographic, and clinical endpoints (Table 13-5). In summary, the totality of evidence appears to point to the similar performance of SES and PES stents in routine de novo coronary artery lesions despite a lower amount of neointimal hyperplasia with SES as assessed by IVUS and angiography.64–67 Among pooled analyses of more complex or “higher restenosis risk” lesion subsets, there may be a clinical benefit to the Cypher SES because of its more potent suppression of neointimal hyperplasia; but validation of these findings would require large-scale, adequately powered clinical trial before any definitive conclusions could be drawn.68 Concomitant with the large amount of clinical trial data comparing SES, PES, and BMS have come a number of analyses amalgamating trial data across clinical studies to increase overall sample size. In particular, these studies have attempted to address one of the prominent limitations of individual DES studies: the limited power to detect differences in low-frequency safety endpoints, including death, MI, and stent thrombosis. By aggregating data across trials, these DES metanalyses have also served as simple means of summarizing the large number of clinical studies in the published and nonpublished domains. In the largest and most comprehensive metanalysis of DES versus BMS (including 9470 patients from 22 randomized trials and 182,901 patients from 34 observational studies), the use of DES in randomized trials was associated with comparable rates of mortality and MI, with significant reductions in TVR compared with BMS.33 In another analysis, Stettler and colleagues incorporated comparative data from SES versus BMS trials, PES versus BMS trials, and SES versus PES trials in a statistical “network” of trials to discern treatment effects across all included trials.69 In this analysis of 38 trials, including data from 18,023 patients, TLR was lower with SES and PES compared with BMS, with similar mortality among patients treated with SES, PES, and BMS. In this analysis, a reduction in the hazard of MI was observed with SES compared with both BMS (hazard ratio [HR] 0.81, 95% credibility interval 0.66–0.97, P = 0.030) and PES (HR 0.83, 0.71–1.00, P = 0.045). In addition to the Stettler network metanalysis, numerous analyses have focused on the examination of low-frequency safety endpoints in comparing first-generation DES with BMS. More than 50 nonrandomized comparisons between DES and BMS have been published and/or presented. Aside from the initial publication of data from the SCAAR (Swedish Coronary Angiography and Angioplasty Registry) study, the majority of these studies have demonstrated favorable safety for DES compared with BMS. For example, in the largest such analysis of DES safety among 262,700 Medicare beneficiaries in the United States, the use of DES was associated with lower rates of death (13.5% vs. 16.5%, P < 0.001) and MI (7.5% vs. 8.9%, P < 0.001) as well as minimal differences in overall revascularization or bleeding.70 Similar findings were observed in a metanalysis of observational studies (not including the Medicare analysis), with reductions in death, MI, and TLR seen with DES compared with BMS.33 Despite the findings from numerous observational registries suggesting that DES may indeed reduce mortality compared with BMS, data from these nonrandomized comparisons of DES versus BMS should be considered exploratory at best and potentially misleading. This observation is based upon several factors: (1) nonrandomized treatment comparisons are subject to significant confounding, which cannot be adequately accounted for using conventional statistical methodology; (2) mortality reductions have never been observed in randomized trials comparing first-generation DES with BMS31; and (3) in propensity-matched observational analyses comparing DES to BMS, the majority of benefit of DES compared with BMS was evident within the first 30 days after implantation—a difference that does not have an adequate pathophysiological explanation.71 These limitations notwithstanding, the abundance of randomized trial and registry study data with first-generation DES has been reassuring, demonstrating the efficacy of both SES and PES in reducing clinical restenosis, and with no major safety concerns compared to BMS.
 Second-Generation Drug-Eluting Stents
Second-Generation Drug-Eluting Stents
Everolimus-Eluting Stents (Xience V/Promus)
In the EES (manufactured by Abbott Vascular, Santa Clara, CA and distributed as the Xience V stent—also distributed by Boston Scientific as the Promus stent)—everolimus (100 mcg/cm2) is released from a thin (7.8-µm), nonadhesive, durable, biocompatible fluorocopolymer consisting of vinylidene fluoride and hexafluoropropylene monomers coated onto a low-profile (81 mcg strut thickness) flexible cobalt chromium stent. The release kinetics are similar to those seen with sirolimus from the SES (~80% of the drug released at 30 days, with none detectable after 120 days). The polymer is elastomeric and experiences minimal bonding, webbing, or tearing upon expansion. Fluoropolymers have been demonstrated to resist platelet and thrombus deposition in blood-contact applications.72,73 The polymer has also been demonstrated to be noninflammatory in porcine experimental models. The low-profile stent struts facilitate rapid reendothelialization74 and are fracture-resistant. Preclinical studies have demonstrated more rapid coverage of the stent struts with functional endothelium with EES compared with SES, PES, or ZES.47 In the small SPIRIT First trial, the EES was shown to markedly reduce the extent of angiographic late loss at 6 and 12 months compared with the otherwise identical cobalt chromium Vision BMS.75 Subsequently, the safety and efficacy of the EES has been compared with that of the PES, SES, and ZES in 15,426 patients enrolled in 11 randomized clinical trials36,76–87 (Table 13-6). Compared with the PES, the EES has been consistently shown to have lower angiographic late loss and restenosis, resulting in less recurrent ischemia, TLR, and TVR. In addition, the EES has been shown to reduce the early and late rates of stent thrombosis and MI compared with the PES. In the large-scale SPIRIT IV trial,36 3,687 patients with stable coronary artery disease undergoing PCI of up to three lesions in three vessels were randomized to the EES versus the PES (Express platform). Patients with unstable acute coronary syndromes, MI, thrombus, chronic occlusions, vein graft lesions, and true bifurcation lesions were excluded. The primary endpoint of target lesion failure (TLF)—a composite of cardiac death, target-vessel MI, or ischemia-driven TLR—at 1 year was reduced by 39% (3.9% vs. 6.6%, P = 0.0008). EES compared with PES also reduced the 1-year rates of stent thrombosis (0.3% vs. 1.1%, P = 0.003), MI (1.9% vs. 3.1%, P = 0.02), and TLR (2.3% vs. 4.5%, P = 0.0008). These results were sustained at the 2-year follow-up.83 In the COMPARE trial,88 1,800 unrestricted “all comers” patients were randomized to EES versus PES (Liberté platform). The primary endpoint of MACE at 1 year (death, MI, or TVR) was reduced by 31% with the EES (6.2% vs. 9.1%, P = 0.02), driven by reductions in stent thrombosis (0.7% vs. 2.6%, P = 0.002), MI (2.8% vs. 5.4%, P = 0.007) and TLR (1.7% vs. 4.8%, P = 0.0002). Between 1 and 2 years in this high-risk study cohort (in whom only ~15% of patients were maintained on dual antiplatelet therapy), fewer stent thrombosis, MI, and TLR events occurred with EES compared with PES.79 In contrast to the marked differences between EES and PES, smaller differences have been found between EES and SES in four randomized trials (Table 13-6). In the ISAR-TEST-4 trial,76 1,304 patients were randomized to EES versus SES, with routine follow-up angiography planned in all patients at 6 to 8 months and 2 years (an additional 1,229 patients were also randomized to an investigational combination sirolimus/probucol-eluting stent, the results of which are not considered here). EES compared with SES at 2 years resulted in lower rates of binary restenosis (12.7% vs. 16.9%, P = 0.03) and ischemia-driven TLR (9.9% vs. 13.5%, P = 0.06), with nonsignificant differences in death, MI, and stent thrombosis. In the SORT OUT IV trial,78 2,774 unselected patients in Denmark were randomized to EES versus SES and followed through the Danish Civil Registration System and Western Denmark Heart Registry. The primary 9-month endpoint of the composite of cardiac death, MI, definite stent thrombosis, or TVR occurred in a comparable proportion of patients in both groups, although the event rates were lower than expected. Definite stent thrombosis occurred in fewer EES than SES patients (0.1% vs. 0.7%, P = 0.05). Similarly, in the EXCELLENT trial,80 late loss was comparable with EES and SES, and the 9-month MACE rates were extremely low in both groups, although there tended to be fewer stent thromboses with EES (0.4% vs. 0.8%, P = NS). Finally, in the ESSENCE-Diabetes trial,86 EES compared with SES resulted in lower rates of angiographic late loss and binary restenosis at 8 months in randomized patients with diabetes. In summary, in a broad cross section of patients undergoing PCI, EES have shown marked improvements in safety and efficacy outcomes compared with PES and more modest improvements compared with SES. Long-term follow-up is still ongoing in most of these trials, the results of which are necessary to determine the robustness of these findings. Moreover, larger randomized trials (with careful monitoring) are required to determine with confidence the magnitude of the differences in low-frequency events between different stents eluting rapamycin analogues. The EES has been compared with the Resolute ZES in one large randomized trial,87 as described below and in Table 13-6.
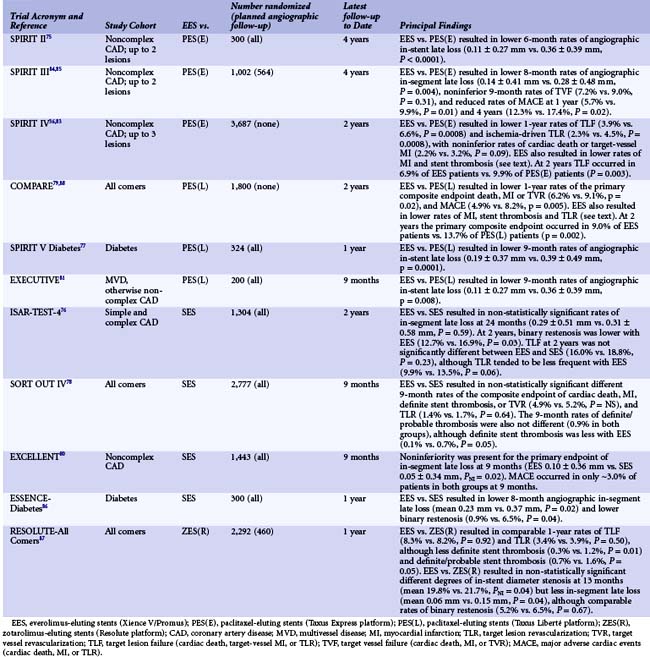
Zotarolimus-Eluting Stent I (Endeavor)
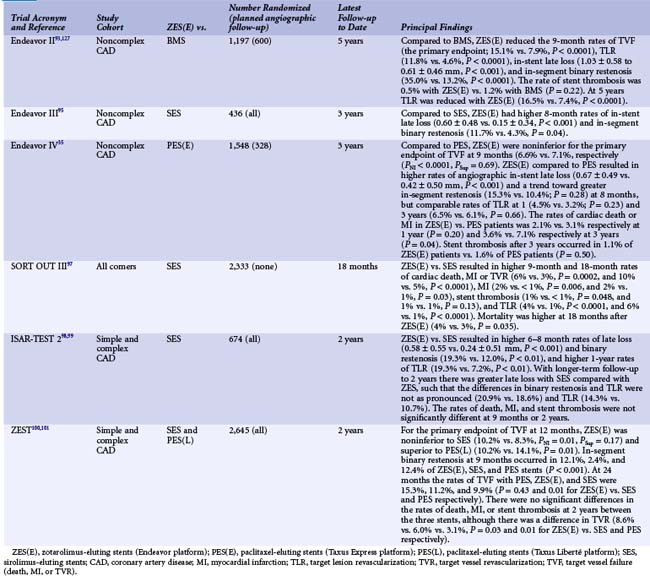
The Endeavor stent (Medtronic, Inc., Santa Rosa, CA) elutes zotarolimus (10 mcg per 1 mm stent length) from a thin layer (5.3 µm) of the biocompatible polymer phosphorylcholine from a flexible, low-profile (91 µm strut thickness) cobalt chromium stent. Phosphorylcholine is a naturally occurring phospholipid found in the membrane of red blood cells that is resistant to platelet adhesion.89 The potencies of zotarolimus, everolimus, and sirolimus are roughly comparable, and zotarolimus is somewhat more lipophilic. However, the release rate of zotarolimus from Endeavor (~90% within 7 days, 100% within 30 days) is significantly faster than the rate at which everolimus and sirolimus are released from Xience V/Promus and Cypher stents respectively. In the Endeavor I FIM study,90 the zotarolimus-eluting Endeavor stent (ZES[E]) was implanted in 100 patients with noncomplex coronary lesions. Although TLR was required in only 1% of patients at 1 year, mean in-stent late lumen loss was 0.33 mm at 4 months and 0.61 mm at 12 months. While significantly less than the late mean in-stent loss of 0.8 to 1.0 mm typically experienced with BMS, this degree of late loss would be expected to result in greater rates of binary restenosis and TLR compared with DES such as EES and SES, which achieve more potent suppression of neointimal hyperplasia.91,92 The ZES(E) was subsequently compared with an otherwise identical BMS in the ENDEAVOR II trial93,94 in 1,197 randomized patients with noncomplex coronary artery disease. As described in Table 13-7, compared with the BMS, the ZES(E) resulted in significantly reduced rates of TVF and TLR at 9 months, which were sustained for 5 years. Although the 9-month rate of angiographic in-stent late loss was 0.61 ± 0.46 mm, this was sufficient to reduce in-segment binary restenosis to 13.2% from 35.0% with the BMS (P < 0.0001). A series of trials were then performed in which the ZES(E) was compared with other DES. In the ENDEAVOR III trial, the rates of late loss and restenosis were significantly greater with the ZES(E) than the SES.95 With 436 randomized patients, this study was not powered for clinical endpoints. In contrast, the ENDEAVOR IV trial (n = 1,548) demonstrated that despite greater late loss and angiographic restenosis with the ZES(E) compared with the PES, the ZES(E) had noninferior 9-month rates of TVF and comparable 12-month rates of TLR in patients with noncomplex coronary lesions.35 In addition, with follow-up through 3 years, significant differences have emerged in the rates of cardiac death or MI favoring the ZES(E).96 Notably, within 1 year, a trend was present for more stent thrombosis (ARC [Academic Research Consortium] definite or probable) with the ZES(E) than the PES. Between 1 and 3 years, however, there was only 1 episode of stent thrombosis with the ZES(E) versus 11 such events with the PES (Fig. 13-6), representing a very favorable late safety profile. Three investigator-sponsored randomized trials have been completed in which the ZES(E) was compared with other DES in unrestricted patient populations. In the SORT OUT III trial,97 2,333 unselected patients (nearly 50% of whom presented with acute coronary syndromes) were randomized at five Danish centers to ZES(E) vs. SES. The 9-month primary endpoint of MACE (cardiac death, MI, or TVR) occurred significantly more frequently with the ZES(E)s (6% vs. 3%, P = 0.0002). ZES(E) also resulted in higher 9-month rates of MI, stent thrombosis, and TLR, differences that persisted at 18 months (Table 13-7). The ISAR-TEST-2 trial was a three-way 1 : 1:1 randomized trial in 1,007 patients of an investigational combination sirolimus/probucol-eluting stent versus the ZES(E) and the SES.98,99 Angiographic follow-up was performed at 6 to 8 months and 2 years. The 674 patients randomized to the ZES(E) had higher rates of late loss, angiographic restenosis (the primary endpoint), and TLR at 6 to 8 months than those assigned to the SES, with similar rates of death, MI, and stent thrombosis (Table 13-7). Greater neointimal hyperplasia and more frequent TLR were evidenced with the SES than the ZES(E) between 6 to 8 months and 2 years, however, such that at the end of 2 years the differences were not as marked. Finally, the ZEST investigators randomized 2,645 patients with simple and complex coronary artery disease to the ZES(E), SES, or PES(L).100,101 Angiographic follow-up at 9 months demonstrated less late loss and binary restenosis with the SES than the other two stents, with lower rates of TVR and TVF with the SES at 1 and 2 years (Table 13-7). The results with the ZES were intermediate between the SES and the PES(L). There were no significant differences in the 2-year rates of death, MI, or stent thrombosis between the two stents.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


