Chapter 26
Atherosclerotic Risk Factors
General Considerations
Christos Liapis, John Kakisis
The word atherosclerosis is derived from two Greek roots: αθηρι (athere, gruel) and σκληρóς (skleros, hard). In 1755, von Haller first applied the term atheroma to a common type of plaque that, on sectioning, exuded yellow pultaceous content from its core.1 In 1904, Marchand introduced the term atherosclerosis in acknowledgment of the consistent association of fatty degeneration and arterial stiffening.2
Atherosclerosis is a systemic disease of large and medium-sized arteries in which lipid and fibrous material accumulate within the intimal layer. It should not be confused with the more general term arteriosclerosis, which refers to generalized thickening and loss of elasticity of arteries as a result of an increased amount of basement material and plasma protein deposition.3
Advances in our understanding of the pathogenesis of atherosclerotic vascular disease gave birth to the concept of cardiovascular risk factors, initially referred to in the Framingham Heart Study.4 Risk factor assessment is important to accurately guide primary and secondary prevention, whereas compliance with risk factor modification is still an area of concern.
Some risk factors are hereditary (and therefore not controllable), but others are acquired or related to behavioral and environmental factors and thus potentially amenable to manipulation. More recently, new risk factors have emerged.
This chapter summarizes the interaction of risk factors with the arterial wall during the atherosclerotic process and briefly reports the currently considered major risk factors. The emerging role and potential management of minor risk factors and markers of atherosclerosis are reviewed. Finally, the importance of compliance with these strategies, including knowledge that appeared in the literature since the publication of the seventh edition of the textbook, is discussed.
Risk Factors and Atherogenesis
The interaction of risk factors with the arterial wall initiates the atherosclerotic process (Fig. 26-1). The starting point for atheroma formation is endothelial dysfunction (reported also as “activation” because cells frequently respond normally to a noxious stimulus).5,6 Chapter 5 covers the mechanisms of atherogenesis in detail; this chapter provides a summary of the key mechanisms related to individual risk factors.
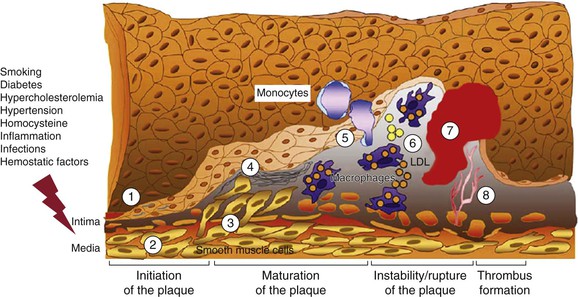
Figure 26-1 Evolution of arterial wall changes and plaque formation in the response-to-injury hypothesis: 1, endothelial dysfunction; 2, vascular smooth muscle cell hypertrophy; 3, migration and proliferation of vascular smooth muscle cells; 4, matrix elaboration; 5, expression of adhesion molecules and migration of monocytes; 6, uptake of low-density lipoprotein (LDL) and formation of foam cells; 7, thrombus formation; 8, angiogenesis and neovascularization. (Modified from Defraigne JO: Development of atherosclerosis for the vascular surgeon. In Liapis CD, Balzer K, Fernandes e Fernandes J, Benedetti-Valentini F, editors: Vascular surgery, New York, 2007, Springer, p 24.)
Interaction with Endothelial Cells: Endothelial Dysfunction
Currently, the most important contributors to endothelial dysfunction are hemodynamic disturbances, hypercholesterolemia, and inflammation. Etiologic culprits also include cigarette toxins, homocysteine, and a wide spectrum of infectious agents. Inflammatory cytokines (e.g., tumor necrosis factor [TNF]) can also stimulate the expression of proatherogenic genes in endothelial cells.7,8
The response of the endothelium to these stimuli can be rapid. For instance, forearm vascular reactivity increases substantially in the 4-hour period after ingestion of a fatty meal.9 Conversely, low-density lipoprotein (LDL) lowering reduces vascular reactivity.9
Experimental models of tobacco exposure, diabetes mellitus, hypercholesterolemia, and hypertension are characterized by common endothelial abnormalities, such as increased generation of oxidative stress and reductions in bioactivity or synthesis (or both) of endothelium-derived nitric oxide, which results in attenuation of vascular tone.10–14
Chronic endothelial injury eventually leads to endothelial dysfunction and increased permeability (Fig. 26-2). Concomitant events include LDL oxidation and accumulation in the subendothelial space of the intima.15 Oxidized LDL has a proinflammatory and proatherogenic effect, and it has recently been suggested that several of the neoepitopes generated during oxidation are highly immunogenic and result in the generation of autoantibodies.16
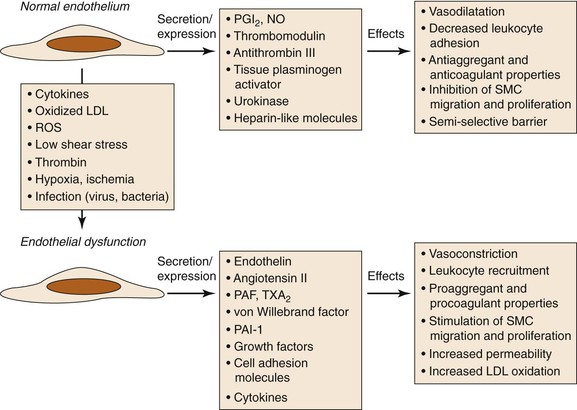
Figure 26-2 Consequences of endothelial dysfunction. Normal endothelium displays antiaggregant, anticoagulant, and vasodilatative properties along with inhibition of cell proliferation. After exposure to various agents causing endothelial dysfunction, these functions are modified toward procoagulant and vasoconstrictive activities together with stimulation of cell recruitment and proliferation. LDL, Low-density lipoprotein; NO, nitric oxide; PAF, platelet-activating factor; PAI-1, plasminogen activator inhibitor-1; PGI2, prostacyclin; ROS, reactive oxygen species; SMC, smooth muscle cell; TXA2, thromboxane A2. (Modified from Defraigne JO: Development of atherosclerosis for the vascular surgeon. In Liapis CD, Balzer K, Fernandes e Fernandes J, Benedetti-Valentini F, editors: Vascular surgery, New York, 2007, Springer, p 25.)
The greater endothelial elaboration of oxygen-derived free radicals activates oxidant-sensitive transcriptional proteins such as nuclear factor κB (NF-κB), which induces the expression of adhesion molecules and thus initiates the inflammatory process.17–22
Both cell recruitment into the endothelial surface and oxidized LDL stimulate the production of growth factors and chemotactic agents by endothelial cells.23 These substances attract and stimulate the proliferation of both macrophages and vascular smooth muscle cells (VSMCs).
Macrophages
As monocytes are attracted to the endothelium and migrate to the subendothelial space, they mature into macrophages and upregulate pattern recognition receptors, including scavenger receptors and Toll-like receptors. Scavenger receptors mediate internalization of oxidized LDL, which results in foam cell formation (the earliest event in the formation of fatty streaks). Toll-like receptors transmit activating signals that lead to the release of cytokines, proteases, and vasoactive molecules. Activated macrophages attract additional monocytes and stimulate VSMCs in a positive feedback loop. As foam cells accumulate in the subendothelial space, they distort the overlying endothelium and may eventually even rupture through the endothelial surface.23
Accumulating evidence indicates that macrophages are responsible for some key features of vulnerable plaque. An intense infiltration of macrophages is invariably observed at the site of plaque rupture, where the fibrous cap appears to be undermined. These macrophages are synthesizing cathepsins and abundant amounts of matrix metalloproteinases (MMP-1, MMP-3, and MMP-9). MMPs degrade the extracellular matrix, which weakens the fibrous cap.24–26
T Lymphocytes
T lymphocytes recruited to the intima interact with macrophages and can generate a chronic immune inflammatory state. The T lymphocytes found in atherosclerotic lesions are polyclonal, which indicates that these cells do not develop in response to a single antigen. It is not clear whether the T lymphocytes respond to specific antigens (e.g., bacterial or viral antigens or modified arterial wall constituents and lipoproteins) or are nonspecifically activated by the local inflammatory milieu.7
Nevertheless, T lymphocytes in atherosclerotic lesions recognize antigens and mount helper T cell type 1 responses with secretion of proinflammatory cytokines, which in turn can stimulate macrophages as well as endothelial cells and VSMCs.27
Subtypes of T lymphocytes called regulatory T cells, previously shown to maintain immunologic tolerance, control the development and progression of atherosclerosis. The function of regulatory T lymphocytes is modulated by chemokines and by costimulatory pathways.28,29
Complement
Activation of complement seems to play a role in both initiation of atherosclerosis and acceleration of the disease.30 Complement activation can occur by the classical (antibody dependent), the alternative (antibody independent), the lectin, or the coagulation pathway.31 Whereas complement activation by the classical and lectin pathways may be protective by removal of apoptotic cells and cell debris from atherosclerotic plaques, activation of the complement cascade by the alternative pathway may be proatherogenic and may play a role in plaque destabilization, leading to its rupture and the onset of acute cardiovascular events. In this context, various complement components, including C3, C3a, C4, C5a, and mannose-binding lectin, have served as biomarkers in prospective clinical studies, associated with an increased risk of death, myocardial infarction (MI), or restenosis after percutaneous transluminal angioplasty.31
Platelets
Platelets play an important role in stimulating the progression of atherosclerotic lesions by secreting growth factors and vasoactive substances (e.g., platelet-derived growth factor, transforming growth factor-α, transforming growth factor-β, epidermal growth factor, and insulin-like growth factor-1) after their adherence to the vessel wall in sites of endothelial ulceration.32,33 These substances recruit and stimulate proliferation of VSMCs.
Recently, platelets have been suggested as initial role players in the development of atherosclerotic lesions by recruiting and binding to leukocytes, endothelial cells, and circulating progenitor cells and initiating transformation of monocytes into macrophages. Platelets internalize oxidized phospholipids, express various scavenger receptors that are able to regulate LDL uptake, and promote foam cell formation.34,35 Moreover, platelets have been identified as key effectors of inflammation throughout plaque development through the secretion of several inflammatory molecules, such as CD40L, P-selectin, RANTES, and Toll-like receptors.36 These molecules exacerbate the inflammation and induce the transition from chronic to acute disease, featuring increased instability of the atherosclerotic lesion that results in plaque rupture and thrombosis.36
Vascular Smooth Muscle Cells
The mediators (e.g., platelet-derived growth factor, fibroblast growth factor, transforming growth factor-β, interleukin [IL]–1, and MMPs) released by endothelial cells, macrophages, lymphocytes, and platelets induce a change in the phenotype of VSMCs from the quiescent “contractile” phenotype to the active “synthetic” state. VSMCs in the synthetic state can migrate and proliferate from the media to the intima, where they produce excessive amounts of extracellular matrix (e.g., collagen, elastin, and proteoglycans) that transforms the lesion into a fibrous plaque. VSMCs are also capable of functions typically attributed to other cell types. Like macrophages, VSMCs can express a variety of receptors for lipid uptake and can form foam-like cells, thereby participating in the early accumulation of plaque lipid.
Like endothelial cells, VSMCs can also express a variety of adhesion molecules (e.g., vascular cell adhesion molecule-1 [VCAM-1] and intercellular adhesion molecule-1 [ICAM-1]) to which monocytes and lymphocytes can adhere and migrate into the vessel wall.37 The lesion grows until it is transformed from a fibrous plaque to a complex plaque. Apoptosis, proliferation, and migration of VSMCs are vital to the pathogenesis of atherosclerosis and plaque rupture. VSMCs are the only cells within plaque capable of synthesizing structurally important collagen isoforms.
Currently, the development of therapeutic approaches targeting VSMC migration and proliferation represents an active field of research, beyond drug-eluting stents and balloons that are already standard procedures for the prevention of restenosis.38–40
Vulnerable Plaque
Vulnerable atherosclerotic plaque (high-risk or unstable plaque) is associated with an increased risk of disruption, distal embolization, and vascular events. Vulnerable plaque is an advanced histologic lesion with a large lipid core (filled with lipid and cell debris), thin fibrous cap, ulceration, intraluminal thrombosis, and intraplaque hemorrhage as well as intense infiltration by macrophages and other inflammatory cells.41–44
The most widely used histologic classification of atherosclerotic plaque is the American Heart Association–recommended Stary classification.44 Inflammation plays a key role in the pathogenesis of atherosclerosis. In this process, the immune system and oxidative stress seem to be involved in the initiation, propagation, and activation of lesions in the arterial wall.41–44 Unstable plaque is dominated by inflammatory cells that destroy the fibrous cap and are responsible for endothelial denudation and thrombogenicity of the plaque contents (Fig. 26-3). Rupture depends on the balance between inflammatory cell activity and the VSMC-driven repair process.45 Activated macrophages, T lymphocytes, and mast cells produce a variety of molecules—inflammatory cytokines, proteases, coagulation factors, radicals, and vasoactive molecules—that are expressed in plaque and may modulate remodeling of the extracellular matrix, cell proliferation, and cell death (apoptosis) and thereby ultimately destabilize these lesions.41–44,46
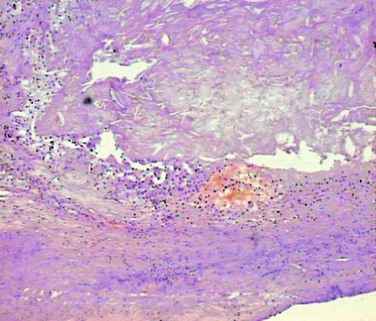
Figure 26-3 Unstable atherosclerotic plaque on histologic examination. Type VI plaque according to Stary’s classification,44 in which one or more of the following are present: surface defects (ulceration), hematoma, and thrombosis.
The likelihood of plaque rupture is a balance between the tensile strength of the plaque and the hemodynamic stress exerted on it.47 In ruptured plaque, the fibrous cap appears to be eroded at the shoulder of the lesion (where the fibrous cap meets the intima of the normal segment of the vessel wall).
Furthermore, local infection (e.g., induced by cytomegalovirus, herpes simplex virus, or Chlamydophila pneumoniae) may influence plaque stability.48
Several invasive and noninvasive imaging methods have been used to identify vulnerable plaque, but all have their limitations (Table 26-1).43 Thus far there are no widely available modalities to adequately provide both anatomic and functional information about vulnerable atherosclerotic plaque. New modalities are on the way, and the future looks promising.49–51
Table 26-1
Advantages and Disadvantages of the Different Imaging Techniques Used for Identification of Vulnerable Plaque
| Imaging Technique | Advantages | Disadvantages |
| High-resolution duplex ultrasound scanning | High resolution; noninvasive; no x-ray exposure; information on arterial wall thickness; qualitative and quantitative analysis of plaque | Limited information on plaque activity, possible improvement with the use of contrast-enhanced ultrasound |
| Digital subtraction angiography | Information on diameter stenosis and luminal surface | Invasive; may underestimate the degree of stenosis; no vessel wall imaging; no information on plaque composition and activity; contrast agent; x-ray exposure |
| Magnetic resonance angiography | Noninvasive; no x-ray exposure; no contrast agent; information on plaque composition | Less accurate in severe stenosis (unless contrast is used) |
| Helical computed tomography angiography | Noninvasive; differentiates between soft, intermediate, and calcified plaque | Contrast agent; x-ray exposure; overestimation of stenosis; no information on plaque activity |
| Intravascular ultrasound | Characterizes vessel wall and plaque morphology early in the disease process; differentiates stable and unstable plaque | Invasive; no information on plaque activity |
| Positron emission tomography | High sensitivity; functional information (plaque activity); molecular imaging | Lack of anatomic information unless combined with computed tomography; radiation exposure |
| Single-photon emission computed tomography | High sensitivity; functional information (plaque activity); molecular imaging | Lack of anatomic information unless combined with computed tomography; radiation exposure |
| Intravascular thermography | Functional information (plaque activity) | Invasive; risk of endothelial damage; difficult interpretation |
| Optical coherence tomography | High resolution (fibrous, lipid, and calcified components of the plaque can be distinguished); small probe size | Invasive; needs blood displacement; limited time window for imaging (2 seconds); limited length of penetration (1-2 mm) |
| Elastography | Differentiates lipid-rich and fibrous tissues; high sensitivity and specificity | Invasive |
| Near-infrared spectroscopy | Information on tissue chemical composition; detects the lipid core, the fibrous cap, and inflammation | Influenced by flowing blood; lack of anatomic information; limited independent role |
Assessment and Management of Risk Factors
The prevalence plus severity of atherosclerosis and peripheral arterial disease (PAD) or coronary artery disease (CAD) in individuals and groups is related to several “old” and “new” risk factors. Thus far there has been no universal agreement on the exact classification of the various cardiovascular risk factors and risk markers.
Classification of Risk Factors
An American Heart Association prevention conference statement in 199952,53 classified risk factors into three categories: “traditional”/conventional risk factors, which appear to have a direct causal role in atherogenesis; predisposing factors, which mediate some risk through the causal factors but may also have independent effects; and conditional risk factors, which have “an association with an increased risk for CAD, although their causative, independent, and quantitative contributions to CAD are not well documented.” The authors suggested that these factors may enhance risk in the presence of causative risk factors, hence the term “conditional.”
Since then, extensive research in all clinical fields of atherosclerosis has identified several novel and emerging risk factors or “markers” of atherosclerosis, which, however, need further confirmatory studies. As soon as sufficient evidence is available, some of them will move to the category of conditional risk factors, but it remains to be elucidated which ones will ever prove to be predisposing or even conventional.
A contemporary classification of risk factors is presented in Box 26-1.5,52–58
Conventional Risk Factors
Conventional risk factors for atherosclerosis are similar in the limbs and in the carotid, coronary, and other vascular beds. The four major risk factors for vascular disease59–65 are discussed in detail in other chapters of this section—smoking (see Chapter 27), diabetes mellitus (see Chapter 28), hyperlipidemia (see Chapter 29), and hypertension (see Chapter 30)—and are not discussed here.
Predisposing Risk Factors
Advanced Age
Although atherosclerosis may be present in younger individuals, age has a dominant influence. All forms of cardiovascular disease become more prevalent in the elderly. In several studies, the risk for PAD increased 1.5- to 2.0-fold for every 10-year rise in age,59,66–68 whereas the prevalence of intracranial atherosclerosis increases by 5% for every 1-year rise in age.69 A recent systematic review of 40 studies showed that the prevalence of carotid artery disease among people younger than 70 years was 4.8% in men and 2.2% in women, rising to 12.5% and 6.9%, respectively, among those 70 years of age and older.70 Similarly, death rates from CAD increase with each decade of life up to the age of 85 years. The death rate from CAD in white men aged 25 to 34 years is about 10 per 100,000; by the age of 55 to 64 years, it has increased 10-fold to nearly 1000 per 100,000.71
Obesity
Obesity predisposes patients to the development of hypertension, diabetes mellitus, and hyperlipidemia. Excess adiposity has recently been shown to play a direct role in initiating atherosclerosis in that fat cells are capable of affecting the systemic vasculature through a variety of mechanisms, among which altered arterial homeostasis, endothelial dysfunction, and inflammation are the most prominent.72–74 Weight loss interventions modify not only the risk factors but also arterial function.
In the Cardiovascular Health Study, each 5-unit increase in body mass index in midlife and in older age was associated with about a 30% increase in PAD prevalence and incidence.75 The European Prospective Investigation into Cancer and Nutrition (EPIC) study showed that general (as indicated by the body mass index) and abdominal (as indicated by the waist circumference or the waist-to-hip ratio) adiposity are both associated with the risk of death.76 Abdominal adiposity is a better predictor of death in patients with a low body mass index. The underlying mechanism is that adipose tissue, especially adipose tissue from visceral fat deposits, secretes mediators that are important in the development of chronic diseases, such as respiratory ailments, cancer, and atherosclerosis. In support of this theory, the risk of death from cardiovascular disease or cancer had been found to be higher among patients with a higher body mass index.77
Physical Inactivity
It is well established that the absence of even moderate levels of physical activity increases risk for cardiovascular morbidity and mortality in the whole population. It is hypothesized that physical activity consisting of either structured training or self-controlled body motivation results in “pleiotropic” actions on the cardiovascular system, which is explained, at least in part, by the modification of traditional and novel cardiovascular risk factors (e.g., hypertension, dyslipidemia, obesity, insulin resistance, endothelial dysfunction, hyperglycemia, inflammation, hypercoagulability, and oxidative stress).78–80 However, only 60% of the beneficial effect of physical activity on cardiovascular disease can be attributed to favorable changes in known risk factors, indicating that physical inactivity may be an independent risk factor for cardiovascular disease.80,81 Moreover, a substantial and expanding body of evidence has demonstrated that low exercise capacity is an independent predictor of cardiovascular and overall mortality.82–85 Therefore, lifelong exercise training should be incorporated in all strategies of primary or secondary prevention against cardiovascular diseases.86–89
Gender
Gender influences the risk for atherosclerosis and its preceding risk factors (e.g., hyperlipidemia, hypertension, and insulin resistance). Epidemiology, symptoms, and progression of cardiovascular disease are different between men and women. Indeed, cardiovascular disease develops in women about 10 years later than in men and typically after menopause. Premenopausal (estrogenic) women are relatively protected against atherosclerosis and its consequences compared with age-matched men. Estrogen alters serum lipoprotein levels through estrogen receptor–mediated effects on hepatic expression of apoproteins. With the onset of menopause, LDL levels rise, whereas high-density lipoprotein (HDL) levels fall, thereby changing a previously antiatherogenic lipid profile to one similar to that of men. However, several clinical trials have failed to demonstrate any utility of postmenopausal hormonal therapy for prevention of vascular disease in women.90–92
Family History and Genetics
Most cardiovascular events result from the interactions of genetic and environmental factors, none of which can cause disease by itself.
Familial predisposition to atherosclerosis is multifactorial and related to familial clustering of other risk factors (e.g., hypertension, hyperlipidemia, and diabetes), direct genetic linkage to atherosclerosis, or family habits associated with environmental risk factors.93 After adjustment for known risk factors, familial history of CAD remains an independent predictor of the disease, associated with a twofold to threefold increased risk of CAD.65 Particularly, individuals with a familial history of CAD of early onset (age at onset before 55 years in men and before 65 years in women) should be regarded as a high-risk group.65 Similarly, three family studies have assessed the heritability of the ankle-brachial index and demonstrated that about 20% to 50% of the interindividual variability in the ankle-brachial index is attributable to genetic determinants.94 Although aneurysm development is a combination of atherosclerosis and degeneration, the significant association with family history indicates genetic predisposition.95
Approximately 40 quantitative trait loci for atherosclerotic disease have been found in humans. Linkage analysis has identified specific genes that may contribute to cardiovascular risk, for example, myocyte enhancer factor-2 (MEF2A) with risk for MI96 and arachidonate 5-lipoxygenase–activating protein (ALOX5AP) with risk for MI and stroke.97 In femoral artery samples from patients with PAD, 366 genes were differentially regulated in intermediate lesions and 447 in advanced lesions.98 Among the upregulated genes, several genes involved in immune and inflammatory responses were identified.
In the future, a complete identification of genetic markers of atherosclerosis may be used in clinical prediction algorithms and even in therapeutic strategies. Resources such as the Human Genome Project and the international HapMap project are expected to change the practice of cardiovascular medicine in the 21st century.99
Behavioral and Socioeconomic Factors
Poor community socioeconomic conditions, depression, social isolation, work- or family-related stress, and type A behavioral patterns have been correlated with CAD and the severity and progression of atherosclerosis. These psychosocial risk factors are conveyed by behavioral pathways, such as inappropriate diet, smoking, sedentary lifestyle, and inadequate use of medical resources, and psychobiologic mechanisms, such as disturbed autonomic and hormonal regulation, which is directly involved in the pathogenesis of cardiovascular diseases.100–104
The risk for angina and ischemic stroke is two to four times higher in blacks than in whites, whereas the risk for PAD is highest in non-Hispanic blacks,105,106 not explained and not related to diabetes, hypertension, or body mass index.107
Conditional Risk Factors
Conditional risk factors had initially been proposed to facilitate classification of factors that may enhance the risk for cardiovascular disease in the presence of causative risk factors. Even though evidence for these factors has improved toward “independency,” additional prospective studies and randomized trials are needed to provide level A evidence.5,52–58
Homocysteine
Homocysteine is an intermediate metabolic product in the transmethylation and transsulfuration reactions that are essential for normal cell growth, differentiation, and function. It is involved in the transformation of methionine to cysteine.108 Plasma levels of homocysteine are regulated by environmental factors, such as vitamin B levels, as well as by genetically determined factors, such as the rare cystathionine β-synthase deficiency and enzyme activity of 5,10-methylenetetrahydrofolate reductase.109
Reports suggest that elevated homocysteine levels are an independent risk factor for vascular disease.110 Elevated homocysteine is associated with a mildly increased risk for CAD, stroke, PAD, and venous thromboembolism with a risk ranging between 0.8 and 3.4.111–113 In a meta-analysis of studies that adjusted for six or seven Framingham risk factors, each 5-µmol/L increase in homocysteine level conferred an approximately 9% increase in the risk for cardiovascular events.114
Homocysteine exerts its atherothrombotic effect through several potential mechanisms. It enhances thrombosis by forming a less porous fibrin network; by activating platelet aggregation and procoagulant factors V and VII, thrombomodulin, tissue factor pathway inhibitor, and plasminogen activator inhibitor (PAI); and by inhibiting activation of protein C. It also triggers proinflammatory activity by activating NF-κB and by promoting monocyte differentiation through NAD(P)H oxidase–mediated oxidant stress, endothelial dysfunction by inhibition of nitric oxide availability by endothelial cells and formation of reactive oxygen species, the LDL oxidation usually detected in cases of atherosclerotic lesions, proliferation of smooth muscle cells, and expression of tissue factor in endothelial cells and macrophages.115–119
Plasma levels of homocysteine are usually regulated by supplementation with B vitamins, either dietary or prescribed as folate.117,120 However, several clinical trials have demonstrated that lowering of homocysteinemia with vitamin supplementation is not effective in reducing cardiovascular risk,121–123 and even a harmful effect from combined B vitamin treatment has been suggested.124
C-Reactive Protein
C-reactive protein (CRP) is an acute phase protein. It is primarily synthesized by hepatocytes, driven by IL-6 with synergistic enhancement of IL-1 or TNF.125 A rise in CRP levels by as much as 1000-fold is not uncommon in both infectious and noninfectious disorders, including MI.126 Until recently, it has been the primary inflammatory marker used in clinical practice, and its predictive value can be estimated only through high-precision assays (high-sensitivity CRP [hsCRP]), with acceptable precision down to or below 0.3 mg/L (2.86 nmol/L). It is within these lower, previously “normal” ranges that hsCRP levels seem to have predictive ability for cardiovascular events. An hsCRP level higher than 10 mg/L (95.24 nmol/L), for example, should be discarded and measurement repeated in 2 weeks to allow the acute inflammation to subside before retesting.
Observational as well as prospective studies have consistently reported that elevated CRP serum levels definitely have prognostic value for cardiovascular events and mortality. CRP has been suggested to induce a prothrombotic state by induction of tissue factor expression in human monocytes.127 It can activate or inhibit the complement system, driving the inflammation in atherosclerotic lesions.128 CRP has also been reported to decrease the expression and bioactivity of endothelial nitric oxide synthase,129 with a subsequent effect on vasodilatation. CRP inhibits both basal and vascular endothelial growth factor–stimulated angiogenesis, whereas it promotes endothelial apoptosis in a nitric oxide–dependent fashion.130 CRP has been found to synergistically enhance angiotensin II–induced proinflammatory effects, such as cellular migration and proliferation, and lesion collagen and elastin content.131 CRP induces the release of monocyte chemotactic protein-1 (MCP-1) and endothelin-1 and thereby upregulates adhesion molecules and chemoattractant chemokines in endothelial cells and VSMCs.132 Finally, CRP has been suggested to mediate the proliferation and activation of VSMCs through activation of NF-κB,133 which leads to accumulation of these cells in the vascular intima and initiates atherogenesis.
Several therapeutic strategies orientated toward a reduction in CRP have been proposed, including cyclooxygenase inhibitors (aspirin and cyclooxygenase 2 inhibitors), angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and especially statins.134 In a randomized clinical trial comprising 17,802 apparently healthy women and men with LDL cholesterol levels of 130 mg/dL or lower and hsCRP levels of 2.0 mg/dL or higher, LDL cholesterol levels were reduced by 50% and hsCRP levels by 37% in patients randomized to 20 mg daily rosuvastatin instead of placebo.135 After a median follow-up of 1.9 years, the rates of most endpoints, including MI, stroke, arterial revascularization, and unstable angina, were reduced by about 50%, whereas all-cause mortality was reduced by 20%. Given the short follow-up time, the beneficial outcome of rosuvastatin may reflect the plaque stabilization effect of lowering CRP levels rather than the inhibition of bulky progression of existing plaque.
However, the causal association between CRP and vascular disease has been challenged by other studies showing that polymorphisms in the gene encoding CRP are associated with marked increases in CRP levels but are not in themselves associated with an increased risk of ischemic vascular disease.136 Similarly, morphometric analyses of atherosclerotic plaques in CRP-deficient animals revealed equivalent or increased atherosclerotic lesions compared with controls, an experimental result that does not support a proatherogenic role of CRP.137
Fibrinogen
Fibrinogen is a glycoprotein that circulates at high concentration in blood. Fibrinogen initially mediates platelet aggregation. Later in clot formation it is converted to fibrin. The fibrin matrix gives the clot shape, strength, flexibility, and stability.
Levels of fibrinogen have been demonstrated to be elevated in patients with acute thrombosis. Fibrinogen increases plasma viscosity and induces VSMC proliferation. The Gothenburg Heart Study was the first prospective trial to show an association between fibrinogen levels and subsequent risk for cardiovascular disease.138 In the Northwick Park Heart Study,139 fibrinogen level appeared to be as effective as total cholesterol concentration in predicting future risk for CAD. Higher levels of fibrinogen predict subsequent acute coronary syndromes, whereas lower levels, despite elevated cholesterol levels, are associated with a lower risk for acute coronary syndromes.140 It remains unclear, however, whether elevated fibrinogen levels are a cause or a consequence of atherosclerosis. In the Copenhagen City Heart Study, the relative risk of stroke development was almost double in patients with higher fibrinogen levels. Nevertheless, elevated levels were not associated with echolucent unstable carotid plaque.141 On the contrary, the CARDIA (Coronary Artery Risk Development in Young Adults) study demonstrated that an elevated fibrinogen concentration in persons aged 25 to 37 years is independently associated with subclinical cardiovascular disease (as indicated by the prevalence of coronary artery calcification and increased mean carotid intima-media thickness) in the subsequent decade.142
Modification of fibrinogen levels has failed to reduce the overall risk for MI or sudden cardiac death.143
Emerging Factors and Markers of Atherosclerosis
Inflammatory Markers
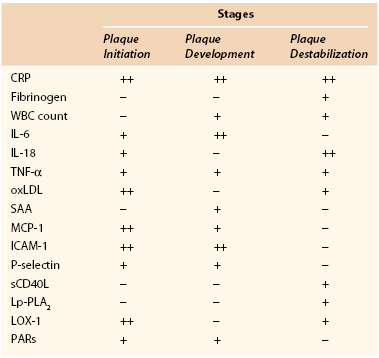
Apart from CRP and fibrinogen, several other inflammatory markers have been associated with atherosclerosis and cardiovascular disease (Table 26-2).
Serum Amyloid A.
Serum amyloid A is an acute-phase reactant, an apolipoprotein associated with HDL, that is predominantly produced by the liver. Nevertheless, it is also implicated in chronic inflammatory diseases (rheumatoid arthritis, atherosclerosis). Serum amyloid A has been shown to have a number of potentially proatherogenic effects that can lead to the destabilization of an atherosclerotic plaque: promotion of chemotaxis for monocytes and neutrophils; stimulation of the production of other proinflammatory cytokines, such as IL-1β and TNF-α; reduction of reverse cholesterol transport; and induction of the MMPs.144 Serum amyloid A has also been shown to promote thrombosis by increasing tissue factor, leading again to the destabilization of atherosclerotic plaques.144 Serum amyloid A was found to be marginally associated with cardiovascular disease,145 especially with extension of CAD in women.146
White Blood Cell Count.
In peripheral blood, the white blood cell count is usually increased in inflammatory and infectious conditions and could be affected in plaque inflammation as well. A higher leukocyte count has been associated with a greater cardiovascular risk. In a meta-analysis of seven prospective studies comparing the top with the bottom third of the value distribution, the relative risk of coronary disease was 1.4 (95% CI, 1.3 to 1.5),147 thus rendering leukocytes a valuable marker. Consistent with this, the decrease in the leukocyte count, achieved by pravastatin in patients with CAD, has been found to be an independent predictor of inhibition of the progression of coronary atherosclerosis.148
Cytokines.
Cytokines are key regulatory glycoproteins allied to inflammatory and immunologic processes that modulate all aspects of vascular inflammation. They are intimately associated with atherogenesis and modulate plaque morphology and stabilization. Many cytokines have been implicated in atheroma formation and complication. Several studies have established the role of IL-6 as an independent predictor of PAD in community screening, irrespective of ethnicity.149–151 IL-6 enhances cell adhesion molecule expression and the production of acute-phase reactants such as CRP and TNF-α by hepatocytes. Exogenous administration of IL-18 to mice enhances atherosclerotic lesions,152 whereas inhibition of IL-18 results in a reduction in atherosclerosis.153 IL-18 has a bearing on the progression and stability of human atherosclerotic plaque.154 TNF-α is involved in the progression of atherosclerosis from the initial stages of intimal thickening to the subsequent vessel occlusion. It stimulates expression of selectin and adhesion molecules and production of MMP-1, MMP-9, MMP-11, and MMP-13 in the endothelium, VSMCs, and macrophages.155 Locally within the atheroma, it increases expression of tissue factor, a potent thrombogenic protein.156 Finally, MCP-1 is the most widely studied chemokine in PAD and CAD,157 and it is consistently expressed in both animal and human atherosclerotic plaque.
Endothelial Adhesion Molecules.
Induction of endothelial adhesion molecules (ICAM-1, P-selectin) is considered one of the earliest steps in atherogenesis. Adhesion of leukocytes to the vascular endothelium and subsequent migration into the intima are key events in the atherosclerotic process. Several cytokines induce expression of ICAM-1, VCAM-1, and selectins by endothelial and smooth muscle cells and promote leukocyte adhesion at the site of the vascular lesion.158
Circulating Soluble CD40 Ligand.
Largely derived from activated platelets, circulating soluble CD40 ligand (sCD40L) can trigger an inflammatory reaction in vascular endothelial cells by the secretion of cytokines and chemokines. Interaction of membrane-bound CD40L and sCD40L with the CD40 receptor leads to the release of matrix MMPs and subsequent destabilization of the plaque.159 Elevated plasma concentrations of sCD40L at baseline predict a subsequent increased risk for future cardiovascular events in apparently healthy women and stable angina patients.160
Protease-Activated Receptors.
Members of the G protein–coupled receptors, protease-activated receptors mediate the cellular effects of thrombin. Protease-activated receptors contribute to the proinflammatory phenotype observed in endothelial dysfunction, and their upregulation in VSMCs seems to be a key element in the pathogenesis of atherosclerosis and restenosis.161
Lipoprotein-Associated Phospholipase A2.
Lipoprotein-associated phospholipase A2 is a proinflammatory enzyme secreted by macrophages and leukocytes. Its activity is associated with a risk for CAD and ischemic stroke.162
Because of their involvement in the atherosclerotic process, some of the inflammatory markers became important therapeutic targets. However, trials using TNF-α antagonists have failed to show any improvement in cardiac function.163,164 Statins have been found to have an independent effect on reducing MCP-1 levels.165 Nevertheless, no marked differences in TNF-α levels have been demonstrated in PAD patients receiving statin therapy.166 Peroxisome proliferator–activated receptor-γ agonists may inhibit production of cytokines and expression of adhesion molecules in endothelial cells167 and reduce production and entry of MMPs into plaque.168 Overall, the clinical value of anticytokine therapies remains to be proved.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


