Chapter 29
Atherosclerotic Risk Factors
Hyperlipidemia
Teresa L. Carman, Carl Orringer
Based on a chapter in the seventh edition by Roger Walcott and William James Howard
Clinical evidence has established the role of atherogenic lipoproteins in the pathogenesis of atherosclerotic vascular disease and the benefits of lipid management for the primary prevention and improvement of existing atherosclerotic vascular disease.
Although much of the evidence on the management of lipid disorders has resulted from studies on atherosclerotic cardiovascular disease, and to a lesser extent, cerebrovascular disease, this generalized experience is also applicable to the prevention and treatment of peripheral artery disease (PAD) because of the common pathophysiology of atherosclerosis in all vascular beds. Available studies of lipid management in patients with PAD demonstrate the positive benefits of recognizing and managing atherogenic lipoproteins in this patient population. Multisocietal practice guidelines for the diagnosis and treatment of PAD developed by the American College of Cardiology (ACC) and the American Heart Association (AHA) published in 2005 recognize that coronary heart disease (CHD) and PAD share the common risk factors of hypertension, dyslipidemia, diabetes, and smoking.1 Furthermore, these guidelines recommend a reduction in low-density lipoprotein (LDL) cholesterol levels to the same therapeutic goals recommended for CHD. It is increasingly recognized that focusing pharmacologic efforts on LDL reduction alone may not be sufficient for atherosclerotic management. Non–high-density lipoprotein (non-HDL) cholesterol levels or apolipoprotein B (apo B) levels may provide a better estimate of atherogenic cholesterol (Box 29-1).
In this chapter, we review the metabolism of lipoproteins, with emphasis on the atherogenic lipids. We discuss the diagnosis of the lipid disorders of concern for vascular surgeons. Clinical trials of lipid management in patients with PAD are presented as well as the clinical trial evidence that established the current guidelines, therapeutic goals, and treatment options of these lipid disorders. Both lifestyle and pharmacologic therapy for atherogenic lipid disorders are reviewed.
Physiology and Metabolism of Lipids
Lipids are hydrophobic and are chemically insoluble in blood. They are transported as lipoproteins in which the hydrophobic lipid components are surrounded by an envelope of hydrophilic phospholipids and proteins known as apolipoproteins (apo). This creates a water-soluble spherical particle that can be carried in the blood. The principle function of lipoprotein particles is transporting lipids from the intestine and liver through the bloodstream to the cells of the body, where they can be stored, used for synthetic processes, and metabolized to yield energy. The various apolipoproteins, such as apo B, apo A, apo E, and apo C, serve as cofactors in metabolism of the contained lipid and as ligands to facilitate binding of lipoproteins to receptors on the surface of cells throughout the body.
Chylomicrons
Lipoprotein metabolism can be divided into exogenous and endogenous phases.2 During the exogenous phase, both dietary lipids and re-circulated lipids within bile are absorbed into enterocytes and packaged as very large lipoproteins called chylomicrons. Although dietary cholesterol and triglycerides are absorbed by different mechanisms within the gastrointestinal (GI) tract, they are combined in this single chylomicron particle for transport from the GI tract through the thoracic duct and into the circulation to the rest of the body. Triglycerides constitute approximately 90% of the chylomicron’s lipid content. When present in significant concentration, chylomicrons account for the turbidity or “milkiness” of plasma, known as postprandial lipemia, which is seen in some individuals with metabolic and inherited disorders of lipid metabolism. Chylomicrons are distinguished by the presence of one apo B-48 molecule in each particle. In the bloodstream, the enzyme lipoprotein lipase (LPL) hydrolyzes the triglyceride contained within the chylomicron into free-fatty acids (FFAs). FFAs are taken up by peripheral tissues and used for energy. In adipose and muscle cells, FFAs can also be re-esterified into triglycerides and stored for future energy production. The remaining triglyceride-poor chylomicron remnant contains only the absorbed dietary cholesterol, which is then transported to the liver for storage. Although chylomicrons are typically considered a transport lipoprotein, evidence suggests atherosclerotic lesions contain apo B-48 within plaque, thus implicating these chylomicron remnants as atherogenic particles.3,4
Very-Low-Density Lipoproteins
The endogenous phase of lipoprotein metabolism involves the hepatic formation of very-low-density lipoproteins (VLDLs) containing cholesterol and triglycerides derived from stores within the liver and adipocytes (Table 29-1). Each VLDL particle contains apos C and E as well as one molecule of apo B-100 per particle. Similar to chylomicrons, the predominant lipid component of VLDL is triglyceride, which accounts for approximately 70% of its lipid content. Although not as large as chylomicrons, VLDL is large enough to cause lipemia when present in very high concentrations. VLDL is released from the liver into the bloodstream, where LPL again facilitates removal of the triglyceride component of VLDL and presents it to the muscle cell as fuel for energy production. Through this endogenous mechanism, triglycerides stored in adipocytes, hepatocytes, and muscle cells can be used for energy during fasting or starvation as a more energy-rich alternative to glucose. As the triglyceride is removed, two additional atherogenic lipoprotein particles are formed: VLDL remnants and intermediate-density lipoproteins (IDLs). These triglyceride-rich lipoprotein particles are atherogenic and play an important role in the accelerated atherosclerosis observed in metabolic syndrome and type 2 diabetes mellitus.
Low-Density Lipoprotein
In addition to LPL, hepatic lipase participates in the conversion of IDL to LDL, the most atherogenic of all lipoprotein particles. Although a number of other apolipoproteins are attached to the original VLDL particle, only one, apo B-100, is present in each LDL particle. LDL binds to specific LDL receptors on the surface of each cell, facilitating transfer of the remaining cholesterol to these cells, where it can be stored and used as cell membranes, steroid hormones, and bile acids. The number of exposed LDL receptors is regulated by the intracellular concentration of cholesterol within each cell. When excess plasma LDL is present, atherosclerosis can possibly increase in proportion to the concentration of circulating LDL.5,6
Brown and Goldstein received the Noble Prize in 1985 for discovering the LDL receptor. They were able to demonstrate that the circulating LDL concentration in plasma is determined by the number of LDL receptors on the various cells of the body, with the liver accounting for more than 70% of this total receptor number.7 When the intracellular cholesterol content of the cells is low, LDL receptor synthesis is upregulated, receptor numbers increase, and the LDL plasma concentration decreases. In contrast, when intracellular cholesterol is increased, LDL receptor synthesis is downregulated, receptor numbers diminish, and circulating LDL rises. Humans are born with a maximum number of LDL receptors and a very low circulating LDL level of 25 to 30 mg/dL (0.65-0.78 mmol/L).8 Over a lifetime, the Western lifestyle of excessive calories, cholesterol, and saturated fat intake, along with inactivity, results in increasing intracellular cholesterol levels, causing secondary downregulation of LDL receptors. Therefore, observed increases in excess plasma LDL cholesterol levels coincide with the epidemic of atherosclerosis seen worldwide.
High-Density Lipoproteins
HDL is synthesized by the liver and intestine as apo A-I, which is released into the bloodstream as a lipid-poor discoid particle (Fig. 29-1). Stored cholesterol released from peripheral cells through the action of a specific transporter known as adenosine triphosphate–binding cassette (ABC) transporter A19 is absorbed by the discoid apo A-I and converted to cholesterol ester under the influence of lecithin-cholesterol acyltransferase (LCAT). During this process, HDL becomes a spherical particle. Additional cellular cholesterol is then added by another cassette transporter, ABCG1, and through the action of the scavenger receptor B1 (SR-B1). The HDL particle can then return to the liver, where it binds to hepatic SR-B1 and releases its cholesterol, or it can exchange a portion of its cholesterol content for triglyceride from VLDL through the chemical action of the cholesterol ester transfer protein (CETP). This removes triglyceride from VLDL, converting it into LDL, which is then removed from circulation through the hepatic LDL receptor. This process is known as “reverse cholesterol transport” and plays an important role in the antiatherogenic properties of the HDL particle.10,11
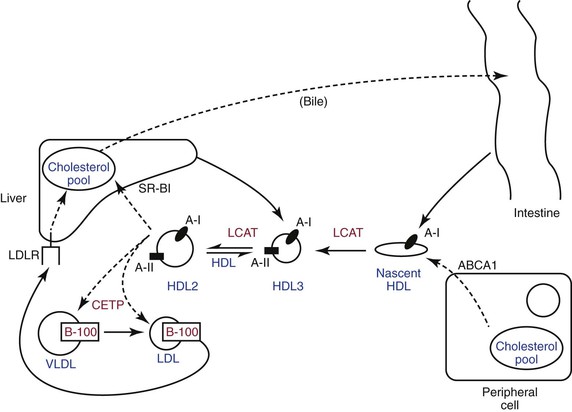
Figure 29-1 High-density lipoprotein metabolism and reverse cholesterol transport. ABCA1, Adenosine-binding cassette transporter 1; CETP, cholesterol ester transfer protein; HDL, high-density lipoprotein; LCAT, lecithin-cholesterol acyltransferase; LDL, low-density lipoprotein; LDLR, LDL receptor; SR-B1, scavenger receptor type B1; VLDL, very-low-density lipoprotein.
Pathophysiology of Atherosclerosis
The pathophysiology of atherosclerosis is deemed to be the same in the peripheral and cerebrovascular circulation as it is in the coronary arteries. To varying degrees, the traditional risk factors are the same, regardless of the location of atherosclerotic disease, including hyperlipidemia, which is both sufficient and necessary for the formation of atherosclerotic lesions throughout the arterial circulation. Under ideal circumstances, the arterial endothelium maintains normal arterial hemodynamics and serves as a barrier to atherosclerosis by preventing circulating atherogenic lipoproteins from entering the arterial wall.
Although atherosclerosis is not fully understood, it develops over time through a complex association between inflammation and endothelial cell injury. Endothelial injury can result from increased lipid levels and from other risk factors, such as hypertension, cigarette smoking, and hyperglycemia. When injured, the endothelium secretes deleterious cytokines that can promote the entry of circulating macrophages into the arterial wall, which stimulates their proliferation and conversion to tissue macrophages with expression of the “scavenger receptor” for LDL particles.12 The endothelium also becomes permeable to LDL and other circulating atherogenic lipoproteins (VLDL remnants, IDL, and chylomicron remnants). Once inside the vessel wall, LDL is oxidized by free radicals, furthering the inflammatory immune response and initiating the pathologic process of atherosclerosis. Oxidized LDL is subsequently absorbed through specific scavenger receptors into fixed tissue macrophages, thereby transforming them into lipid-filled foam cells.13 Groups of foam cells accumulate underneath the endothelium and become the initial lesion of atherosclerosis (the fatty streak). As this process continues, the foam cells undergo apoptosis, or cell death, which allows the lipid contained in them to spill out to form the lipid core of an atherosclerotic plaque.14 Although the arterial wall may initially remodel and expand to maintain the lumen (a process known as positive remodeling) as the plaques continue to grow, become fibrotic, and intrude on the arterial lumen, the lumen becomes narrowed.14 Stress-induced tissue ischemia occurs at the critical point of plaque encroachment, and with further growth, ischemia at rest can be observed. This fibrotic plaque is thought to be stable and is not the cause of the majority of acute clinical events (e.g., myocardial infarction [MI] or stroke), but it undoubtedly plays a significant role in the chronic PAD process. Other plaques accumulate a large lipid core, which induces an intense local inflammatory reaction as this lipid is oxidized, which results in the infiltration of additional macrophages and inflammatory cells. This is often referred to a vulnerable or unstable plaque.15 It usually remains small and does not critically compromise the diameter of lumen. However, it is usually covered by a thin fibrous cap that is prone to ulceration or rupture. When this occurs, a platelet-rich clot rapidly forms on top of the plaque and produces complete obstruction of the involved artery and subsequent acute clinical infarction or ischemia.
Diagnosis of Atherogenic Lipid Disorders
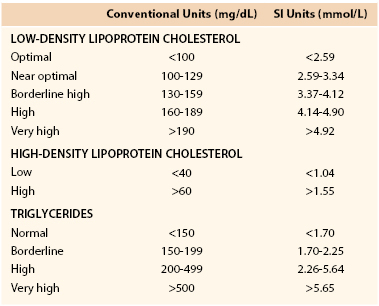
The Adult Treatment Panel III (ATP III) of the National Cholesterol Education Program (NCEP) recommended that all adults older than 20 years be screened for hyperlipidemia every 5 years with a fasting lipoprotein profile.16,17 This study should include total cholesterol (TC), triglycerides, HDL cholesterol, and LDL cholesterol. It is recommended that patients be in their usual state of health and fast for 12 hours before testing. Blood should be drawn after sitting comfortably for 5 minutes and with less than 1 minute of tourniquet time. If a fasting specimen cannot be obtained, TC and HDL cholesterol should be measured as a screening alternative. If TC is greater than 200 mg/dL (5.18 mmol/L) or HDL is less than 40 mg/dL (1.04 mmol/L), a fasting lipoprotein profile should be obtained to better quantify the patient’s atherosclerotic risk.
Fasting Lipid Profile
ATP III recommended new guidelines for the evaluation of a fasting lipid profile (see Table 29-1). TC is not a good risk predictor because it can be increased by elevated HDL, which is usually a lower risk lipid profile, or decreased by low HDL, which is a higher risk lipid profile. The presence of very high triglyceride levels can also elevate TC and suggest a higher risk lipid profile than actually may be present.
LDL cholesterol has been shown to be the most predictive lipoprotein fraction for determining atherosclerosis risk in both epidemiologic and interventional studies, and the associated risk is directly proportional to the LDL cholesterol concentration over a wide range of values.18,19 HDL cholesterol is equally predictive in epidemiologic studies, with an inverse relationship between the HDL cholesterol concentration and risk for atherosclerosis.18,19 The risk associated with each of these lipoprotein fractions is additive in individuals with increased LDL cholesterol and decreased HDL cholesterol.20 Increased triglycerides are more difficult to evaluate for prediction of risk. In univariant analyses of the influence of triglycerides on atherosclerotic risk, elevated triglycerides are associated with an increased risk of atherosclerotic vascular disease.19 However, multivariate analyses of risk suggest triglycerides are less important as a risk predictor due to the co-dependence of elevated triglycerides with other atherosclerotic risk factors such as low HDL, hypertension, and hyperglycemia (i.e., diabetes).20 When very sophisticated analyses of triglyceride-rich lipoproteins are performed,21 it is evident that large chylomicrons and VLDLs are not atherogenic, but the smaller remnant forms of both these lipoproteins and the presence of IDL definitely confer increased atherogenicity.
Clinically, elevated triglycerides in the presence of type 2 diabetes, metabolic syndrome, or familial combined dyslipidemia should indicate an increase in the risk for atherosclerotic cardiovascular disease. Very high levels of triglycerides (>1000 mg/dL [11.3 mmol/L]) are also associated with an increased risk for pancreatitis. Atherogenic dyslipidemia is a characteristic lipoprotein pattern frequently observed in patients with type 2 diabetes mellitus and the metabolic syndrome. In this condition, the LDL cholesterol is often unimpressive or frankly low, but it is associated with elevated triglycerides with a decreased HDL cholesterol concentration. More important, however, is the change in the number and composition of LDL particles associated with the metabolic syndrome and type 2 diabetes, rendering them more atherogenic. This LDL is described as “small, dense LDL,” which is an increased number of particles that contain less cholesterol per particle. Studies have shown that it is the number of LDL particles (LDL-P) that truly reflects the risk related to this profile.22 LDL-P can be determined by lipid analysis using nuclear magnetic resonance (NMR) studies. Because each LDL-P contains one apo B-100 molecule, another method of more accurately assessing the risk conferred by an individual’s LDL profile is to measure the apo B-100 level. Complete analysis may provide a better risk assessment because the individual risks associated with each lipoprotein abnormality in type 2 diabetes mellitus and metabolic syndrome are additive.
Non–High-Density Lipoprotein Cholesterol in Risk Assessment
Non-HDL cholesterol has been recognized in epidemiologic studies as one of the better predictors of atherosclerotic risk.23 Non-HDL cholesterol is easily calculated by subtracting HDL cholesterol from TC. ATP III recommended the use of non-HDL cholesterol (see Box 29-1) as a way to more accurately assess risk in patients with increased triglycerides and made this a secondary target of therapy after the primary therapeutic goal for LDL cholesterol is attained (discussed in the following). Non-HDL cholesterol essentially represents the cholesterol content in all of the apo B–containing atherogenic lipoproteins: non-HDL cholesterol = LDL cholesterol + IDL cholesterol + VLDL cholesterol + lipoprotein (a) (Lp[a]). However, these more specialized studies of cholesterol fractions are significantly more expensive than a routine lipid profile. Therefore, it is recommended that the initial risk evaluation and lipid management be performed with the standard LDL cholesterol, HDL cholesterol, and non-HDL cholesterol measurements and that specialized testing be reserved for unusual situations or for fine tuning of therapy.
Lipoprotein (a)
Epidemiologic studies have established that Lp(a) is associated with increased atherosclerotic risk and risk of cardiovascular events, particularly ischemic stoke.24,25 However, little is known about the metabolism of this hybrid lipoprotein particle. There is a lack of interventional data to establish Lp(a) as a legitimate modifiable atherosclerotic risk factor, and there are few effective therapies for lowering levels of this lipoprotein. For this reason, Lp(a) is categorized by ATP III as an emerging risk factor. When present in increased concentration, it should be considered added evidence for more aggressive lipid management, similar to the presence of other emerging risk factors, such as high-sensitivity C-reactive protein (hs-CRP) and other inflammatory markers. The European Atherosclerosis Society recommends checking Lp(a) levels in patients at moderate or high risk of cardiovascular disease, including patients with a personal or family history of premature atherosclerosis, familial hypercholesterolemia, family history of elevated Lp(a), recurrent atherosclerotic events despite statin therapy and meeting accepted goals for therapy, and patients with increased 10-year risk for major cardiovascular events according to European or U.S. guidelines.25
Evolution of the Guidelines for Management and Therapeutic Goals for Lipoprotein Disorders
Adult Treatment Panel I
The NCEP was launched in 1988. It has served as the driving force for lipid management guidelines since the publication of the ATP I report and recommendations for standardization of clinical measurement of lipids.26 ATP I focused both on public health and primary prevention of CHD in individuals with an increased lifetime risk for CHD because of increased LDL cholesterol. The public health focus was designed to make U.S. healthcare providers aware of the risks associated with lipid risk factors and to encourage screening of the adult population for risk assessment. In addition, ATP I sought to educate the U.S. population regarding CHD risk and to encourage cholesterol reduction. The focus of ATP I was education and lifestyle intervention. Interventional therapy was reserved for individuals at increased risk, including those with an LDL greater than 190 mg/dL (4.92 mmol/L) or those with an LDL cholesterol greater than 160 mg/dL (4.14 mmol/L) and two or more additional CHD risk factors (Box 29-2).
Adult Treatment Panel II
The NCEP published the ATP II report in 1993 after the introduction of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins).27 These guidelines continued the ATP I recommendations and broadened the scope of the NCEP to include secondary prevention of CHD in patients with known CAD. Analysis of several of small clinical trials that used quantitative coronary angiography to evaluate the effect of diet compared with diet and pharmacologic therapy in patients with known CHD demonstrated a significant reduction in progression of angiographic CHD in the active treatment groups compared with progression of CHD observed in patients who received placebo therapy.28 In addition, despite being underpowered for clinic events, a clear trend toward improvement was seen in participants who achieved a significant improvement in their lipid profile. Although the majority of the effect was ascribed to pharmacologic-associated reductions in LDL cholesterol, several of the trials also resulted in significant increases in HDL cholesterol,28 and this lipid change was thought to contribute to the positive outcomes of these surrogate endpoint trials. On the basis of these secondary prevention studies, ATP II recommended that patients with known CHD have the more aggressive therapeutic goal for LDL cholesterol of 100 mg/dL or less. The other recommendations of ATP I for primary prevention remained the same.
A secondary observation was made from the analysis of the coronary angiograms in these trials.28 The plaque that caused significant clinical events was almost always less than 70% obstructing, and clinical events often resulted from plaque that caused a less than 50% reduction of the coronary lumen. This introduced the concept of a “vulnerable plaque.”29 This is a small plaque with a large lipid core and a thin fibrous cap that is prone to rupture or ulceration. Because this plaque typically does not cause critical coronary stenosis and is not accompanied by the formation of collateral circulation, when fully obstructed by the sudden development of a clot on top of the vulnerable plaque, a more serious infarction results. It was suggested that statin therapy “stabilized” this plaque by helping prevent rupture or ulceration of the fibrous cap.
On the basis of these secondary prevention studies, ATP II recommended that patients with known CHD have the more aggressive therapeutic goal for LDL cholesterol of 100 mg/dL or less. The other recommendations of ATP I for primary prevention were continued.
Adult Treatment Panel III
Initially published as an executive summary16 and later as a full report in 2002,17 ATP III was the first evidence-based report from the NCEP that focused on large clinical trial data. Both primary prevention studies, the West of Scotland Primary Prevention Study (WOSCOP)30 and the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS),31 and secondary prevention trials, the Scandinavian Simvastatin Survival Study (4S),32 the Long-Term Intervention with Pravastatin in Ischemic Disease (LIPID) study,33 and the Cholesterol and Recurrent Events (CARE) trial,34 were included in the evaluation that demonstrated the benefit of statin therapy compared with placebo. As a result, ATP III recommended a more aggressive approach to risk assessment and management of lipid levels. Table 29-1 shows the new classification recommended by ATP III for LDL cholesterol, HDL cholesterol, and triglyceride levels in the initial risk assessment of individuals based on lipid levels. ATP III also recommended more aggressive goals of therapy for lipid management in patients with both established CHD and CHD risk equivalents. In addition to continuing the evaluation of additional risk factors (see Box 29-2) recommended by ATP I and II, ATP III added an emphasis on 10-year risk assessment for CHD by using the Framingham risk assessment algorithm.
Coronary Heart Disease Risk Equivalents
Another feature of ATP III was the designation of three conditions as “CHD risk equivalents,” in that each carried a 10-year risk for a major coronary event that was equal to that of patients with established CHD (>20% per 10 years). Diabetes mellitus, other atherosclerotic diseases including PAD, carotid artery disease, and abdominal aortic aneurysm, and the presence of multiple cardiovascular risk factors that conferred a 10-year risk of greater than 20%, as judged by the Framingham risk assessment tool, received the designation of “CHD risk equivalent” (Box 29-3). Although discussed in the ATP II report, ATP III was the first formal recognition of the importance of PAD as a significant indicator of CHD.
Risk Stratification
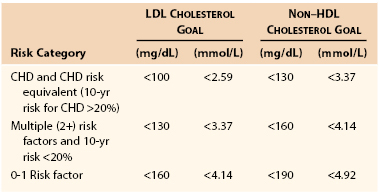
ATP III suggested patients be categorized into one of three risk strata and that aggressiveness matched therapeutics to the assessed risk level. These three categories are as follows: (1) high risk—established CHD and CHD risk equivalents; (2) intermediate risk—multiple (2+) risk factors; and (3) low risk—0 to 1 risk factor. In addition to the goals recommended for LDL cholesterol, in individuals with triglyceride levels greater than 200 mg/dL, a secondary goal was recommended for management of non-HDL cholesterol (discussed previously). Table 29-2 shows the LDL and non-HDL cholesterol goals for the treatment of individuals in each of these three risk categories. Although ATP III introduced the issue of basing lipid management on an individual’s 10-year risk assessment, management of individuals with very high LDL levels or severe individual risk factors that conferred a high lifetime risk was continued as recommended by ATP I and II.
Beyond Adult Treatment Panel III
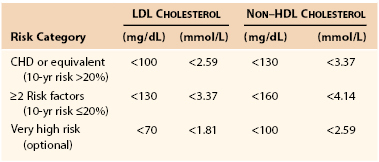
After ATP III was published, trials of very aggressive lipid lowering in very high-risk individuals were conducted, and the LDL cholesterol levels reached were considerably lower than those recommended by ATP III.35,36 The positive clinical outcomes of these aggressive lipid-lowering trials resulted in an addendum to the ATP III report published in 2004 that recommended lower therapeutic goals for LDL cholesterol and non-HDL cholesterol in patients with very high-risk CHD (Table 29-3).37 In 2005, the ACC/AHA published guidelines specifically directed to the management of patients with PAD. Patients with established PAD should be treated with statins to achieve a target LDL of less than 100 mg/dL (class I recommendation). Treatment goals of less than 70 mg/dL is reasonable in patients with lower extremity PAD who are at high risk for events (class IIa recommendation).38 The most recent contribution to evolution of the guidelines for lipid management was the AHA/ACC Guidelines for Secondary Prevention for Patients with Coronary and Other Atherosclerotic Vascular Disease: 2006 Update.39 These guidelines reaffirmed the more aggressive therapeutic goals for LDL cholesterol and non-HDL cholesterol and further recommended that if these goals are not achieved, both LDL cholesterol and non-HDL cholesterol should be reduced by at least 50%. ATP IV guidelines are in development by the National Heart, Lung and Blood Institute (NHLBI) and may be available as early as 2013.40
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


