Chapter 6 Atherosclerosis
Pathology, Pathogenesis, and Medical Management
Vascular surgeons commonly treat patients with the complications of atherosclerosis. Currently, more precise lesion classification and imaging, a better understanding of atherogenesis, and increasingly effective medical treatment before and after vascular interventions promise improved long-term results. A better understanding of atherosclerosis and technical advances for its treatment now provide scientifically based prevention and management strategies. The pivotal role of lipids in the pathogenesis of atherosclerosis has been delineated, along with the effects of treatment on plaques and the recognition of the crucial role of inflammatory and immune responses which affect arterial plaques. Novel risk factors for late progression, independent of conventional risk factors, have been identified; these comprise elevated blood levels of biomarkers such as inflammatory cytokines, metalloproteinases, and smooth muscle growth factors including glucose and insulin. Advanced imaging can detect unstable plaques that are prone to rupture, thrombosis, and downstream embolization. Prospective randomized trials using drugs, micronutrients, and other interventions continue to provide therapeutic guidelines. Coronary thrombosis or stroke, the main causes of death in patients with peripheral arterial disease,1 require active management strategies to improve survival and enhance long-term reconstructive results.
Vascular surgeons must be familiar with the location and natural history of individual lesions and, in considering various interventions, distinguish primary prevention from secondary treatment. When active intervention is required, vascular surgeons will use strategies geared toward the pathology of a specific vascular lesion in particular arterial sites. For example, a stenotic lesion composed of smooth muscle and well-organized collagen, although producing some degree of distal ischemia, is a much safer lesion than a plaque containing an unstable core of atheromatous debris beneath a tenuous cap. The smooth stenosis of an adductor hiatus plaque in the femoral artery, causing stable claudication, is clearly not as threatening as unstable carotid or coronary plaques characterized by soft cores beneath friable caps; these lesions have differing vulnerabilities based on differing composition and morphology.2 It is now recognized that operative or endovascular treatment of segmental lesions does not prevent the progression of systemic atherosclerosis elsewhere. Medical treatment is required to ensure long-term results.
Variations in patterns and rates of progression of atherosclerosis have critical clinical implications for the timing and choice of treatment.3,4 Considering the atherosclerotic process as a single disease leads to oversimplification. With considerable lesion diversity and clinical presentations, atherosclerosis can be viewed as a polypathogenic process comprising a group of closely related vascular disorders.5,6 Multiple risk factors promoting atherosclerosis and its complications include dyslipidemia, smoking, diabetes, hypertension, and proinflammatory factors. This multiplicity of disease-promoting factors make a single-disease–single-etiology view difficult to reconcile. This chapter considers atherosclerosis as though it were a single entity, at the same time recognizing its variable pathology and differing clinical presentations. Despite the complexity of this process, new concepts for treatment have resulted in increasingly favorable outcomes. This progress is based on advances in surgical approaches and effective medical therapy based on a better understanding of the pathology and pathogenesis of this disease.
Pathology
General Concepts
The term atheroma derives from the Greek athere, meaning “porridge” or “gruel”; sclerosis means “induration” or “hardening.” A gruel-like color and consistency and induration or hardening exist to various degrees in different plaques, different disease stages, and different individuals. In 1755, von Haller7 first applied the term atheroma to a common type of plaque that, on sectioning, exuded a yellow, pultaceous content from its core.7 Figure 6-1 illustrates a typical fibrous plaque containing a central atheromatous core with a fibrous or fibromuscular cap, macrophage accumulation, and round cell adventitial infiltration. A past, generic definition of atherosclerotic plaque as “a variable combination of changes in the intima of arteries consisting of focal accumulation of lipids, complex carbohydrates, blood and blood products, fibrous tissue and calcium deposits”8 failed to encompass the spectrum of atherosclerotic lesions. Advanced plaques invade the media, and at certain stages produce bulging or even enlarged arteries. Round cell infiltration, medial changes, and neovascularization characterize advanced atherosclerotic lesions. The atherosclerotic process ultimately involves the entire arterial wall. The process is complex and variable and, although all atherosclerotic lesions may not evolve in the same way, certain patterns of its progression are more common than others.
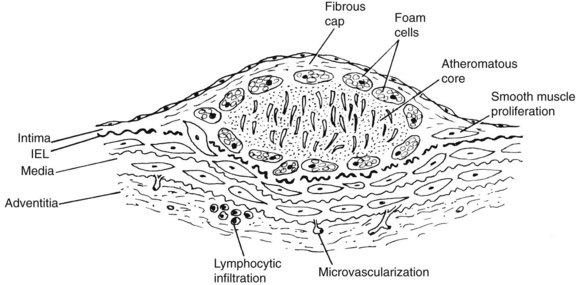
The development and expansion of the lipid atherosclerotic core and its relationship to an overlying cap have been recognized as causes of plaque complications. The observation of a lipid “core,” developing early in atherosclerosis and accumulating in the deep aspects of early lesions before actual fibrous plaque formation begins, is a key insight.9 Another important insight is the recognition of the role of inflammation and immune reactions in the early and late stages of atherogenesis.10,11 The inflammatory cascade includes the appearance of proinflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor (TNF) α, and antiinflammatory cytokines such as IL-10 within arterial tissue and in the bloodstream. Lipid accumulation appears to attract inflammatory cells that produce cytokines locally, whereas cytokines or biomarkers derived from a variety of tissues appear systemically in atherosclerotic subjects. For example, in atherosclerotic claudicants, plasma levels of inflammatory cytokines TNF-α and IL-6 are elevated, whereas antiinflammatory IL-10 levels are reduced.12 Elevated levels of cytokines such as TNF-α and its receptors have also been shown to affect the arterial wall.13–15 The atherosclerotic plaque contains leukocytes, of which approximately 80% are monocytes or monocyte-derived macrophages. Lymphocytes, predominantly memory T cells,16 constitute 5% to 20% of this cell population. Inflammation, size, and composition of the lipid core determine plaque instability or “vulnerability,” promoting sudden expansion, rupture, release of distal emboli, and vascular occlusion.
Fatty Streaks
The first stages of atherosclerosis, fatty streaks, minimally raised yellow lesions, develop in characteristically vulnerable segments of the arterial tree. These lesions contain lipids deposited intracellularly in macrophages and in smooth muscle cells. Stary and colleagues17 defined initial fatty streaks and intermediate lesions of atherosclerosis as follows: type I lesions in children are early microscopic lesions, consisting of an increase in intimal macrophages and the appearance of foam cells. Type II fatty streak lesions are grossly visible; in contrast to type I lesions, type II lesions stain with Sudan III or IV. Foam cells and lipid droplets appear in intimal smooth muscle cells and heterogeneous droplets of extracellular lipids characterize type II fatty streaks. Type III lesions are intermediate lesions, considered to be the bridge between the fatty streak (Figure 6-2) and the prototypical atheromatous fibrous plaque, the type IV plaque (see Figure 6-1).17 Type III lesions occur in plaque-prone locations in the arterial tree19 at sites exposed to forces (particularly low-shear stress) promoting increased low-density lipoprotein (LDL) influx.20
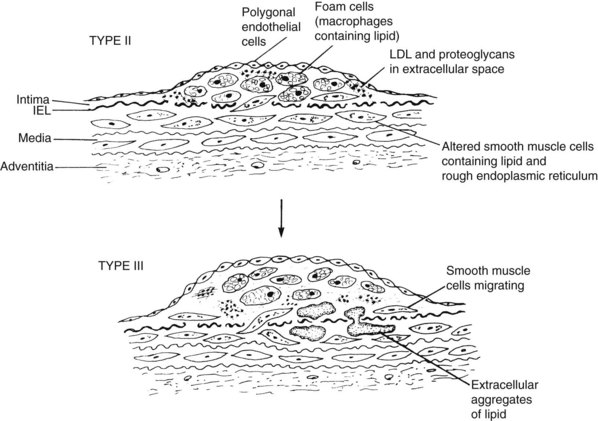
The fatty streak type II lipids are chemically similar to those of plasma.21 Plasma lipids can enter the arterial wall in several ways. As described in a review of pathogenesis,22 LDL accumulation can occur because of (1) alterations in the permeability of the intima; (2) increases in the intimal interstitial space; (3) poor metabolism of LDL by vascular cells; (4) impeded transport of LDL from the intima to the media; (5) increased plasma LDL concentrations; or (6) specific binding of LDL to connective tissue components, particularly proteoglycans in the arterial intima. Experimental studies show that LDL cholesterol accumulates in the intima even before lesions develop and in the presence of intact endothelium. These observations comport with classic descriptions of lesion formation by Aschoff23 in the early twentieth century and Virchow24 in the mid nineteenth century.
A second event in early atherogenesis, as shown in animal experiments, is binding of monocytes to the endothelial lining, with their subsequent diapedesis into the subintimal layer to become tissue macrophages.25–27 Fatty streaks are populated mainly by monocyte-derived macrophages. These lipid-engorged scavenger cells mainly become the foam cells characterizing fatty streaks and more advanced lesions. LDL is altered by oxidation or acetylation to be taken up by the macrophages to form foam cells.28 Oxidized LDL itself is a powerful chemoattractant for monocytes. Another aspect of this theory suggests that the endothelium modifies LDL to promote foam cell formation. In either case, oxidative reactions are seen as enhancing atheroma development.
The initial interactions of plasma LDL with the arterial wall and macrophage appear as the basis for earliest lesion formation. LDL traverses the endothelium mostly through receptor-independent transport, possibly also through cell breaks.29 Endothelial cells,30 smooth cells,31 and macrophages32 are capable of promoting oxidation of LDL. The oxidized LDL, in turn, further attracts monocytes into the intima to promote their transformation into macrophages. Macrophages produce cytokines, which initiates the cytokine inflammatory cascade. Oxidized LDL also induces gene products that are ordinarily not expressed in normal vascular tissue. A notable example is tissue factor, the cellular initiator of the coagulation cascade abundantly expressed by atheroma monocytes and foam cells.33 Expression of tissue factor requires the presence of bacterial lipopolysaccharide, suggesting that hypercoagulability in atherosclerosis might be enhanced by infectious factors.
Large numbers of macrophages and T lymphocytes in the plaques suggest cellular immune responses; oxidized lipoproteins, heat shock proteins, and microorganisms are possible antigens. A study analyzing endarterectomy specimens by immunohistochemistry and reverse transcription polymerase chain reaction showed proinflammatory T cell cytokines, IL-2, and interferon-7 in a large proportion of plaques, indicating that a helper T cell 1–type cellular immune response likely occurs in the atherosclerotic plaque.34
Endothelium
Animal studies reveal that endothelial cells tend to be oriented away from the direction of flow; these cells show increased stigmata or stomata, increased proliferation, and a decrease in microfilament bundles.25,26 In humans and animals, endothelial cells become polyhedral or rounded; in humans, increased formation of multinucleated cells and cilia occurs. Animal studies reveal increased proliferation and cell death, with retraction and exposure of subendothelial foam cells. The endothelium becomes more permeable to macromolecules in experimental models; in humans, it exhibits increased tissue factor expression and mural thrombus formation. Leukocyte adherence increases with the expression of a monocyte adhesion molecule (VCAM-1). Endothelium-derived relaxing factor and prostacyclin release are decreased and vasoconstriction is enhanced.
Macrophages
Macrophages proliferate and express monocyte chemoattractant protein-1, macrophage colony-stimulating factor, TNF, IL-1 and other interleukins, and platelet-derived growth factor (PDGF), along with CD immune antigens and, as previously described, tissue factor.33 Plaque macrophages contain increased free and esterified cholesterol and increased acetyl coenzyme A, cholesterol acyltransferase, and acid cholesterol ester hydrolase. Neutral cholesterol ester hydrolase is decreased. These altered cells also express the scavenger receptor 15-lipoxygenase and exhibit increased lipoprotein oxidation products in humans and in animal models. These extensive changes indicate the complexity of the morphologic, functional, biochemical, and genetic expressions of the arterial wall in early atherosclerosis. The reader is referred to an original report for comprehensive details,17 with references for the cellular alterations given.
Gelatinous Plaques
Intimal gelatinous lesions may, less commonly, be considered atheroma precursors. Haust described these lesions in 1971,35 first noted in 1856 by Virchow24 as potential progenitors of advanced atherosclerosis. Smith later described their identification and composition.36 Gelatinous plaques are translucent and neutral in color, with central grayish opaque areas. Most plaques are characterized by finely dispersed, perifibrous lipid along with collagen strands around the lesions. Grossly, gelatinous lesions feel soft. With gentle lateral pressure, these plaques “wobble.” Gelatinous plaques can be the gelatinous material separates easily from the underlying arterial wall without entering a conventional endarterectomy plane. Gelatinous plaques appear in the aorta as extensive areas of flat, translucent thickenings, particularly in its lower abdominal segment. These lesions have a low lipid and high fluid content. In some plaques, numerous smooth muscle cells are present while the lesions contain substantial amounts of cross-linked fibrin.
Fibrous Plaques
Figure 6-1 typifies the most common prototypical atherosclerotic lesion: fibrous or type IV plaque. These lesions are composed of large numbers of smooth muscle cells and connective tissue, which form a fibrous cap over an inner yellow (atheromatous) core. This soft core contains cholesterol esters, mainly cholesteryl oleate, likely derived from disrupted foam cells. A second type of particle contains both free cholesterol and cholesterol linoleates. The early core is associated with vesicular lipids that are rich in free cholesterol.9 These particles are likely derived directly from LDL, possibly by modification of LDL by specific lipolytic enzymes capable of hydrolyzing LDL cholesterol esters. Lipoprotein aggregation and fusion are thought to be the chief pathway of cholesterol ester accumulation. Fibrous plaques contain large numbers of smooth muscle cells, connective tissue cells, and macrophages. Almost 2 decades ago, the composition and integrity of the atheromatous cap were underscored, as this structure stabilizes the atheroma, preventing intraluminal rupture of its soft core.37
Fibrous plaques appear later than fatty streak and often in similar locations. Fibrous plaques likely evolve from fatty streaks or from areas of fatty streak involvement. Gelatinous plaques, injured arterial areas, or thrombi less commonly may lead to fibrous plaque formation. Mural thrombi can be converted into atheromas, as demonstrated by experimental intraarterial catheter implantation.38
Fibrous plaques protrude into the arterial lumen in fixed cut sections; however, when arteries are fixed at arterial pressure, they produce an abluminal or external bulge. For example, coronary plaques in vivo must occupy at least 40% of the arterial wall before angiographic detection is possible,39 and within limits, atheroma growth is compensated by arterial enlargement.40 Compensatory remodeling of coronary arteries in subhuman primates and humans has been stressed as a protective response.41 However, with lesion growth, ulceration, rupture, or overlying thrombosis, the arterial lumen suddenly becomes compromised, which is a sequence seen in coronary atheromas. A unique adaptive response involving dilatation, with atheromatous involvement of the entire arterial wall and participation of inflammatory cells and immunologically active T lymphocytes, and elastolysis may predispose to aneurysm formation.
During the early stages of evolution from fatty streak to fibrous plaque, cholesterol esters appear in the form of ordered arrays of intracellular lipid crystals. In intermediate type III and fibrous plaques, the lipids assume isotropic forms and occur extracellularly.42 Cholesterol esters and oxysterols are highly irritating, causing severe inflammatory reactions in the connective tissue43; they probably behave similarly within the arterial wall to promote inflammation, fibrosis, and lymphocytic infiltration. Advancing neovascularization from the adventitia characterizes intermediate fibrofatty and fibrous plaque lesions. Atherosclerotic lesions contain immunoglobulin (Ig) G in large quantities, as well as other immunoglobulins and complement components. The IgG recognizes epitopes characteristic of oxidized LDL, indicating that immunologic processes characterize more advanced atherosclerotic plaques.44 This process is associated with systemic effects; for example, patients with carotid atherosclerosis have higher antibody ratios of antioxidized LDL and IgM than do comparable nonatherosclerotic controls.45
Experiments in complement-deficient rabbits suggest that the chronic inflammation of atherosclerosis is driven mainly by activation of the complement and monocyte-macrophage systems.46 In this sequence, enzymatic degradation, not oxidation, is considered to be the central predisposing process.
Complicated Plaques
Fibrous plaques become complicated by calcification, ulceration, intraplaque hemorrhage, or necrosis. These later developments cause the clinical complications of stroke, gangrene, and myocardial infarction. Aneurysm formation can represent a unique genetic or immune interaction with atherosclerosis. Alternatively, aneurysms have been viewed as nonspecific, inflammatory, degenerative, or purely mechanical arterial responses. Patients harboring aortic aneurysms have a high prevalence of risk factors for atherosclerosis and concurrent atherosclerotic involvement of other arteries, suggesting a unique response to atherosclerosis involving this arterial segment in certain individuals.47
As with early plaque evolution,17 advanced atherosclerotic lesions have been described and classified in a separate report.18 The type IV lesion, or atheroma, is potentially symptom producing. Extracellular lipid is the precursor of the core that characterizes type IV lesions. Lesions that contain a thick layer of fibrous connective tissue are characterized as type V lesions, whereas those with fissures, hematoma, or thrombus are characterized as type VI lesions. Type V lesions have been further described as largely calcified (type Vc) or consisting mainly of connective tissue with little or no lipid or calcium (type Vb). This definition of advanced disease includes atherosclerotic aneurysms, though aneurysm formation may follow other distinct sequences.
Theories of Atherogenesis
Lipid Hypothesis
Virchow believed that the cellular changes characterizing atherosclerosis were simply reactive responses to lipid infiltration.24 Later, Aschoff remarked, “From plasma of low cholesterin content no deposition of lipids will occur even though mechanical conditions are favorable.”24 As can be seen from fatty streak to fibrous plaque evolution, lipids, particularly LDL cholesterol, have a pivotal role in lesion morphology, composition, and evolution. Early experiments by Anitschkow with cholesterol-fed rabbits appeared to validate a simple “lipid filtration hypothesis.”48 However, the atherosclerotic process was soon appreciated to be pathogenetically much more complex. Atherosclerosis develops in various species in proportion to the ease with which an experimental regimen displaces the normal lipid pattern toward hypercholesterolemia, particularly hyperbetalipoproteinemia. At the same time, arterial susceptibility and inflammatory responses vary among locations, species, and individuals. Enhanced inflammatory responses, genetically determined by toll-like receptors, are known to influence atherogenesis.49,50
Canine and subhuman primate (rhesus and cynomolgus monkey) models develop atherosclerosis in response to dietary manipulation50–60 and demonstrate plaque regression in response to serum cholesterol lowering. However, lesion production in susceptible species is not a result of simple dietary cholesterol overload. Any diet that causes hypercholesterolemia induces atherosclerosis. The presence of excess, or even any, cholesterol is not necessary in atherogenic diets. In developmental subhuman primate feeding experiments, reduction of cholesterol content to 0.5% combined with sugar and eggs produced rapidly progressive plaques, whereas high cholesterol addition (up to 7% by weight) did not.53,54 In rabbits, a variety of semipure, purified cholesterol-free diets with various amino acid compositions induces hypercholesterolemia and atherosclerosis.60
Epidemiologic observations provide important circumstantial evidence linking hyperlipidemia to atherosclerosis.61 The genetically determined hyperlipidemias provide compelling evidence that elevated LDL cholesterol is a prime etiologic factor in atherosclerosis, despite objections that highly cellular lipid-laden atheromas may be different lesions in these patients.62 These metabolic disorders are most often caused by a lack or abnormality of LDL receptors on hepatocytes, which causes an ability to internalize and metabolize LDL, an important observation that earned Brown and Goldstein a Nobel Prize.63 Serum cholesterol levels are markedly elevated early in life; individuals with the homozygous condition die prematurely from atherosclerosis, rarely living beyond the age of 26 years. Unfortunately, the heterozygous condition is not uncommon, with total cholesterol levels ranging up to 350 mg/dL. These individuals account for 1 in 500 live births64 and develop atherosclerosis during early middle age. The atheromas of these patients are similar in morphology to those seen in individuals with acquired hyperlipidemia or premature atherosclerosis associated with heavy smoking.
This unfortunate natural experiment is powerful evidence that elevated LDL cholesterol is a relentless factor in plaque inception and the rapid progression of atherosclerosis to lethal consequences. Liver transplantation has been successful in retarding the progress of this type of atherosclerosis.65 Familial hypercholesterolemias are autosomal dominant disorders produced by at least 12 different molecular defects of the LDL receptors. Familial abnormalities of high-density lipoprotein (HDL), a negative risk factor for atherosclerosis, also exist. In addition to LDL and HDL metabolism, surface proteins of the lipoprotein complex or apoproteins also appear to be relevant to pathogenesis. With the availability of effective therapy, a need for childhood screening for these disorders has been endorsed recently.66
Thrombogenic Hypothesis
In the mid nineteenth century, von Rokitansky postulated that fibrinous substances deposited on the arterial intimal surface as a result of abnormal hemostatic elements in the blood could undergo metamorphosis into atheromatous masses containing cholesterol crystals and globules.67 This theory held that atheromatous lesions resulted mainly from degeneration of blood proteins (i.e., fibrin deposited in the arterial intima). Duguid68 resurrected this theory in 1946 with the observation that in rabbits, indwelling arterial catheters or arterial injury caused cholesterol accumulation and arterial lesions in the absence of dietary cholesterol.
Mesenchymal Hypothesis: Hemodynamic Effects
Active smooth muscle cells with connective tissue production by these cells have been considered as primary, even crucial steps, in atherogenesis.69–71 Proteoglycan, a ubiquitous arterial wall element, can trap infiltrated LDL, even when LDL is not elevated in the blood. Collagen is the other space-filling component of advanced atherosclerotic lesions. Hauss and colleagues72 viewed the migration of smooth muscle cells from the media to the intima, with proliferation and production of connective tissue, as a nonspecific arterial reaction to any injury; atherosclerosis simply reflects a generic arterial response. Chisolm and colleagues22 called this the “nonspecific” mesenchymal hypothesis. These scenarios are similar to wound-healing responses to injury. In part, this theory attempts to explain why physical factors such as shear stress, vasoactive agents, and repetitive injuries eventually lead to atheroma formation.
In one view, “Atherosclerosis constitutes the degenerative and reparative process consequent upon the hemodynamically induced engineering fatigue of the blood vessel wall.”63 This theory postulates that “the vibrations consisting of the pulsations associated with cardiac contractions and the vortex shedding generated in the blood vessels at branchings, unions, curvatures, and fusiform dilatations (carotid sinus) over a lifetime are responsible for fatigue failure after a certain, but individually variable, number of vibrations.”63 Atherosclerosis, a process of wear and tear, therefore becomes an inexorable (and unavoidable) process associated with aging. In support of this concept, hypertension73 and tachycardia induced in experimental animals receiving atherogenic feeding caused accelerated plaque development, whereas bradycardia induced by sinoatrial node ablation in monkeys reduced coronary and carotid atherosclerosis.74,75
Monoclonal Hypothesis: Smooth Muscle Proliferation
The morphologic similarity of smooth muscle proliferation in some atherosclerotic lesions to uterine smooth muscle myomas led to the suggestion that atherosclerotic lesions are derived from a singular or, at most, a few mutated smooth muscle cells that, like tumor cells, proliferate in an unregulated fashion.70 This theory is based on the finding of only one allele for glucose-6-phosphate dehydrogenase in lesions from heterozygotes. A homology exists between the β chain of human PDGF and the protein product of the v-sis oncogene, which is a tumor-causing gene derived from simian sarcoma virus. Tumor-forming cells in culture express the genes for one or both of the PDGF chains and secrete PDGF into a culture medium. This hypothesis, once again, considers events causing smooth muscle cell proliferation to be a critical atherogenic factor. Actions of other growth factors, which might either stimulate or inhibit cell proliferation, depend on circumstances as well as on macrophage-derived cytokine activity. For example, the finding of transforming growth factor (TGF)-β receptors in human atherosclerosis provides evidence of an acquired resistance to apoptosis.76 Resistance to apoptosis can lead to proliferation of resistant cell subsets associated with progression of lesions.
Response-to-Injury Hypothesis
Ross and Glomset77 initially postulated two pathways for the promotion of atheroma formation. In the first pathway (e.g., in hypercholesterolemia), monocyte and macrophage migration occurs without endothelial denudation. In some instances, endothelial loss occurs, with platelets carpeting bare areas. In this event, platelets would stimulate proliferation of smooth muscle by releasing PDGF.
In the second pathway, the endothelium itself was postulated to release growth factors, stimulating smooth muscle proliferation. Experimental rabbit arterial balloon injury shows that regrowing endothelium induces myointimal proliferation beneath its advancing edges, stimulating accumulation of collagen78 and glycosaminoglycans.79 Stimulated smooth muscle itself then releases growth factors, leading to a continued autocrine proliferative response. In the initial iteration of this theory, the second pathway was postulated to be relevant to atheroma stimulated by diabetes, possibly in relation to insulin-derived growth factors, cigarette smoking, or hypertension. Hypertension causes endothelial injury, but striking differences exist between the behavior of smooth muscle cells in atherosclerosis and hypertension. Atherosclerosis stimulates an overt smooth muscle proliferative response. In most instances, pure hypertension causes thickening of the arterial wall by virtue of increased protein synthesis, without an increase in cell number.80
Arterial trauma, such as clamping or balloon injury, produces stenoses and vascular injuries (ranging from minor to severe) and initiates both myointimal hyperplasia and atheromas. This mode of atherogenesis invokes a response to injury hypothesis. In this scenario, physical or chemical agents cause endothelial denudation, followed by platelet adherence and subsequent release of PDGF,81 triggering smooth muscle migration proliferation and lipid accumulation. This sequence applies to specific arterial wall injuries, particularly when the internal elastic lamina is disrupted.
Injury as a global theory of atherogenesis is unsupported by subsequent observations of early atherogenesis. Arterial denudation is a rare finding in early atherogenesis in humans and animals, although endothelial cells may be injured or dysfunctional while remaining in place.82 It is known that systemic endothelial dysfunction exists in atherosclerosis.83 Endothelial dysfunction exerts profound effects on systemic vasodilatation and the upregulation of endothelial receptors facilitating entry of cells and blood components through an apparently intact endothelium. Because all arterial wall cells secrete growth factors that are similar if not identical to PDGF and its derivatives, postulating physical endothelial disruption and platelet deposition is unnecessary.
The responses of the arterial wall after injury remain of considerable practical interest in both atherogenesis and intimal hyperplasia. With injury, early medial smooth muscle proliferation is the first step, influenced primarily by basic fibroblast growth factor.84 Migration and production of an extracellular matrix are the second and third stages of injury. These mechanisms are relevant to trauma-provoked atheromas, which occur as a result of clamping or balloon injuries in the presence of modestly elevated levels of LDL cholesterol.85 Angiotensin II also causes smooth muscle to proliferate and induces expression of growth factors.86,87 One of these growth factors, TGF-β, exerts either stimulatory or inhibitory effects, depending on circumstances. Injury also induces medial angiotensinogen gene expression and angiotensin receptor expression.88 Other smooth muscle antigens include thrombin, catecholamine, and possibly endothelin. As a result, atheromas developing in a setting of injury (mechanical, immunologic, or infectious) are influenced by trauma-induced growth factors in varying degrees and sequences. However, plasma LDL elevation accentuates neointimal hyperplasia89,90 without actual atheroma formation.
Lesion Arrest or Regression
The potential for plaque regression and stabilization of vulnerable plaques is key in the consideration of prevention and treatment of atherosclerosis. Regression of atherosclerosis in response to lowered serum cholesterol has been demonstrated in autopsy studies of starved humans dating back to World War I,23 in many animal models,91 and in pioneering clinical angiographic trials combining cessation of smoking with lipid reduction.92 In humans, trials of vascular end points have shown some impressive examples of regression in coronary arteries; more commonly, minimal anatomic regression is seen, along with slowing of progression, but with drastic reductions in coronary events.93 Importantly, magnetic resonance imaging has documented favorable longitudinal changes in carotid plaque composition, with reduction of the lipid core and increased fibrous tissue.94
As atherosclerotic plaques in experimental animals regress, plaque bulk is reduced mainly by lipid egress. This has been shown convincingly in experiments using hypercholesterolemic dogs52,53 and monkeys.54–57 The exact mechanisms of lesion regression, particularly the roles of inflammatory and immune responses, are incompletely understood. Regression has been demonstrated using serial observations of decreased bulk of individual plaques; reduced luminal encroachment, as shown by edge defects on sequential angiography; and decreased plaque lipid and altered fibrous protein content measured histologically and chemically.57 An important technical aspect of this research was the confirmation of regressive changes using immediate autopsy or surgical observation and biopsy. Grossly or histopathologically, plaque change correlated with observed regressive angiographic changes.58,59 Stary95 described regression of advanced lesions in atherosclerotic rhesus monkeys, clearly documenting disappearance of macrophages, macrophage-derived foam cells, lymphocytes, and extracellular lipids, as a result of drastic reduction of blood cholesterol for 42 months. Arterial wall calcium deposits, however, did not regress, and as might be expected, calcification is a definite limiting factor in regression and also important while planning endovascular interventions.
Correlative sequential plaque observations are not readily obtained in humans, but angiography and ultrasonography are currently used to assess treatment effects. Experimentally, decreased luminal intrusion on sequential angiography coincided with decreased plaque size and reduced lipid content. In some instances, it has been found that plaque fibrous protein increased during regression.57 Although residual fibrosis might limit plaque bulk reduction, fibrosis may convert a soft atheromatous plaque into a more stable lesion. Active lesions, particularly in the coronary arteries, are not necessarily the most occlusive ones. Although angiographic edge changes appear to be minimal, the reduction in coronary events in response to lipid-lowering treatment likely relates more to plaque stabilization than to bulk reduction. To produce regression consistently, total serum cholesterol must be reduced to approximately 150 mg/dL. Modest serum cholesterol elevations exist above which lesions inevitably progress.59 Antiplatelet treatment does not appear to retard plaque progression; combinations of antiplatelet agents, high-dose aspirin, and dipyridamole in the rhesus model caused rapid, dramatic plaque progression during hypercholesterolemia.60 Favorable lipid thresholds for human regressive responses approximate a total serum cholesterol level of 150 to 170 mg/dL and an LDL level of 100 mg/dL or less, levels that Roberts cited in populations in which atherosclerosis is virtually absent.96 This observation led him to conclude that elevated lipid levels are the single etiologic risk factor in atherogenesis. However, in extrapolating this concept to secondary treatment, inflammatory responses need to be considered. These responses likely contribute to plaque instability leading to atherosclerotic complications, as often occurs, in the absence of hyperlipidemia.
Medical Management
General Considerations
Populations free of coronary disease generally exhibit total cholesterol levels less than 150 mg/dL and LDL cholesterol levels less than 100 mg/dL. This observation questions the primacy of other atherosclerotic risk factors that commonly manifest in these relatively disease free populations.96 In examining the usefulness of the lipid hypothesis for treatment rather than prevention, considerable positive evidence has accumulated to support an energetic approach to overall lipid reduction. The Heart Protection Study—a randomized, placebo-controlled trial using simvastatin in more than 20,000 individuals—showed a reduction of adverse cardiovascular events and prolongation of life when this agent was used for primary and secondary prevention, even in individuals without elevated lipid levels.97 Statins have antiinflammatory effects, as shown by decreased C-reactive protein levels,98 occurring independently of LDL reduction.99,100 Overall, many more individuals are candidates for treatment to achieve recently revised goals of the National Cholesterol Education Project of total cholesterol less than 150 mg/dL and LDL less than 100 mg/dL.101 These stringent lipid target levels can seldom be achieved with diet alone. The Heart Protection Study has recently addressed the issue of baseline C-reactive protein (CRP) in statin therapy, to report, once again, that allocation to simvastatin therapy, proved significantly effective regardless of baseline levels of CRP and in participants with normal lipid levels.102 Empirically a statin, along with low-dose aspirin, clopidogrel (an antiplatelet agent), and angiotensin-converting enzyme (ACE) inhibitors are recommended for patients with peripheral arterial disease.103
Complications and deleterious clinical events associated with atherosclerosis are neither singular nor univariate, but multiple and interactive. In the late stages, the instability of the fibrous plaques involves more than lipid dynamics. More than 4 decades ago, Holman and colleagues104 pointed out that “a sharp line of distinction exists between atherogenesis and the subsequent evolution of lesions that may or may not precipitate clinical disease, for the factors involved in the evolution of lesions beyond the stage of fatty streaks may be entirely different from the factors that initiate fatty streaks.” Among these factors are inflammatory and immune mechanisms, altered fibrous proteins, accumulation of blood elements, and cap rupture. Among interventions intended to produce plaque stabilization or regression, decreasing LDL cholesterol promotes favorable changes in atheromas and improved outcomes. The antiinflammatory statin effect, as judged by biomarkers, also presents as an important synchronous effect. The effect of cigarette smoking, a crucial promoter of complications of atherosclerosis, might be viewed as a contributor to inflammation, which cannot be overcome solely by lipid reduction.
In angiographic regression trials, the most favorable plaque changes in terms of arrest or regression relate to the degree and duration of blood lipid reduction. In peripheral arteries, regression and stabilization were 1.5-fold to twofold more common in treated subjects than in those receiving placebos.92 Although angiographic studies show plaque regression trends, wall change or plaque reduction is usually modest compared with what is believed to be stabilization of vulnerable lesions. Intravascular ultrasound is an important tool to visualized plaque relationships to the arterial wall in response to treatment. Serial angiography has been used to describe favorable clinical results among patients randomly assigned to an experimental group consuming a 10% fat, 12-mg cholesterol diet and undergoing smoking cessation, stress management training, and exercise.104 After 1 year, 82% of the treated group showed regressive changes in coronary artery plaques that depended, in some degree, on the amount of initial lesion encroachment.
Atherosclerosis is often segmental; bypassing or removing symptomatic arterial lesions in selected arterial segments minimizes the deleterious effects of dangerous lesions. These observations, made more than 40 years ago, were uniquely surgical insights and brought life- and limb-saving interventions to many patients.3 Arterial interventions, which include endovascular approaches, evolved as highly effective means of treating patients with advanced, symptomatic atheromas, including specific patterns of coronary involvement, high-grade carotid lesions, and aortic disease. However effective, surgical or endovascular treatment of one arterial segment does not prevent disease progression in other segments––life expectancy remains shortened. Continued smoking after reconstruction make matters worse, particularly after ill-advised infrainguinal or aortic reconstruction for stable claudicants. Aspirin, urokinase, and anticoagulants can prevent or minimize superimposed embolic phenomena and clotting, but underlying plaques continue to progress. Modification of inflammatory responses provoked by cytokine-derived or immune-modulating factors has potential and may now be practical, given the current ability to monitor blood levels for biomarkers such as inflammatory cytokines and CRP, which predict cardiovascular complications and mortality.105,106
Apparently insignificant or small plaques, particularly in coronary or cerebral arteries provoke arterial spasm. Atherosclerotic plaques impair the normal action of endothelium-derived relaxing factor,107–109 impairing vasodilator responses in coronary and cerebral arteries.108 Dietary treatment of experimental atherosclerosis restores endothelium-dependent relaxation responses,110 whereas long-term inhibition of nitric oxide synthesis by feeding promotes experimental atherosclerosis.111
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


