Chapter 8 Atherosclerosis
Concepts of the pathobiology of atherosclerosis have likewise undergone perpetual revision. During much of the 20th century, most considered atherosclerosis a cholesterol storage disease. Recognition of the key role of interactions of vascular cells, blood cells (including leukocytes and platelets), and lipoproteins challenged this model later in the 20th century.1 Current thinking further broadens this schema, incorporating an appreciation of the global metabolic status of individuals, extending far beyond traditional risk factors as triggers to the atherogenic process.
Risk Factors for Atherosclerosis: Traditional, Emerging, and Those on the Rise
Traditional Risk Factors for Atherosclerosis
Cholesterol
Experimental data have repeatedly shown a link between plasma cholesterol levels and the formation of atheromata. Pioneering work performed in Russia in the early 20th century showed that consumption by rabbits of a cholesterol-rich diet caused formation of arterial lesions that shared features with human atheromata.2 By mid-century, application of the ultracentrifuge to analysis of plasma proteins led to the recognition that various classes of lipoproteins transported cholesterol and other lipids through the aqueous medium of blood. Multiple epidemiological studies verified a link between one cholesterol-rich lipoprotein particle in particular—low-density lipoprotein (LDL)—and the risk for coronary heart disease.3 The characterization of familial hypercholesterolemia as a genetic disease provided further evidence linking LDL cholesterol levels with coronary heart disease. Heterozygotes for this condition had markedly elevated risk for atherosclerotic disease. Individuals homozygous for familial hypercholesterolemia commonly develop coronary heart disease within the first decade of life.
Elucidation of the LDL-receptor pathway and that mutations in the LDL receptor cause familial hypercholesterolemia provided proof positive of LDL’s role in atherogenesis.4 Yet the cholesterol hypothesis of atherogenesis still encountered skepticism. Many critics—some laypersons and some respected professionals—questioned aspects of the theory, pointing out that dietary cholesterol levels did not always correlate with cholesterolemia. Lack of proof that either dietary or drug intervention could modify outcomes dogged proponents of the cholesterol hypothesis of atherogenesis.5
Ultimately, controlled clinical trials that lowered LDL by interventions including partial intestinal bypass, bile acid–binding resins, and statin drugs showed reductions in coronary events and vindicated the cholesterol hypothesis. In appropriately powered trials conducted with sufficiently potent agents, lipid lowering also reduced overall mortality. Yet the very success of these interventions suggested there must be more to atherogenesis than cholesterol because a majority of events still occurred despite increasingly aggressive control of LDL cholesterol levels. Identification of proprotein convertase subtilisin/kexin type 9 (PCSK9) as the gene involved in autosomal dominant hypercholesterolemia has furnished new insight in this regard. Reduced function of this enzyme raises cellular LDL receptor numbers, and hence augments LDL clearance, leading to lower plasma LDL levels. Individuals with reduced function variants of PCSK9 who experience lifelong lower exposure to LDL show protection from atherosclerotic events even in the presence of other cardiovascular risk factors. These observations strengthen the case for involvement of LDL in atherogenesis, and for aggressive management of LDL in practice.6
Of course, aspects of the lipoprotein profile other than LDL can influence atherogenesis (see later discussion).3 Yet, because atherosclerotic events commonly occur in individuals with average levels of the major lipoprotein classes, a full understanding of atherogenesis requires consideration of factors other than blood lipids.
Systemic arterial hypertension
The relationship between arterial blood pressure and mortality emerged early from actuarial studies. Insurance underwriters had a major financial stake in mortality prediction. A simple measurement of blood pressure with a cuff sphygmomanometer powerfully predicted longevity. Data emerging from the Framingham Study and other observational cohorts verified a relationship between systemic arterial pressure and coronary heart disease events.7 Concordant observations from experimental animals and epidemiological studies bolstered the link between hypertension and atherosclerosis.
As in the case of high cholesterol, clinical evidence that pharmacological reduction in blood pressure could reduce coronary heart disease events proved fairly elusive. Early-intervention studies readily showed decreases in stroke and congestive heart failure endpoints following administration of antihypertensive drugs. Studies indicating clear-cut reductions in coronary heart disease events with antihypertensive treatment have accumulated much more recently.8
Mechanistically, antihypertensive drug therapy likely benefits atherosclerosis and its complications principally by lowering blood pressure, although some have posited other beneficial actions of various antihypertensive agents. A large randomized clinical trial, the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), showed no advantage over a 5-year period of a thiazide diuretic over an angiotensin receptor blocker, a calcium channel antagonist, or a β-adrenergic blocking agent.9,10
Cigarette smoking
Tobacco abuse, and cigarette smoking in particular, accentuates the risk of cardiovascular events.11 In the context of noncoronary arterial disease, cigarette smoking appears particularly important. The rapid return toward baseline rates of cardiovascular events after smoking cessation suggests that tobacco use alters the risk of thrombosis as much or more than it may accentuate atherogenesis per se. Classic studies in nonhuman primates have shown little effect of 2 to 3 years of cigarette smoke inhalation on experimental atherosclerosis in the presence of moderate hyperlipidemia.
Both first-hand and second-hand tobacco smoke impairs endothelial vasodilator functions—an index of arterial health.12 Smoking has many other adverse systemic effects, including eliciting a chronic inflammatory response implicated in atherothrombosis.13 Cigarette smoking seems to contribute particularly to abdominal aortic aneurysm formation. The mechanistic link between cigarette smoking and arterial aneurysm formation may resemble that invoked in the pathogenesis of smoking-related emphysema. Studies in genetically altered mice that inhale tobacco smoke have delineated a role for elastolytic enzymes such as matrix metalloproteinase (MMP)-12 in the destruction of lung extracellular matrix (ECM). Smoke-induced inflammation appears to release tumor necrosis factor (TNF)-α from macrophages that can elevate activity of elastolytic enzymes and promote pulmonary emphysema.14 A similar mechanism might well promote destruction of elastic laminae in the tunica media of the abdominal aorta, which characterizes aneurysm formation.
Age
Multiple observational studies have identified age as a potent risk factor for atherosclerotic events.15 Indeed, in the current cardiovascular risk algorithm based on the Framingham Study, age contributes substantially to risk calculation. Demographic trends portend a marked expansion in the elderly population, particularly women, in coming years. Although age-adjusted rates of cardiovascular disease may appear stable or even declining in men, the actual burden of disease in the elderly will increase because of their sheer number. In view of the expanding elderly population, evidence that supports the mutability of atherosclerosis assumes even greater importance (see later discussion).
Sex
Male sex contributes to heightened cardiovascular risk in numerous observational studies. Mechanisms for this increased burden of disease may reflect male-related proatherogenic factors and/or lack of protection conferred by female sex. Cardiovascular risk increases after menopause in women; many previously attributed the vascular protection enjoyed by premenopausal women to estrogen. But estrogen therapy in women (in more recent large-scale clinical trials) and in men (in the older Coronary Drug Project study) seems to confer hazard rather than benefit in the circumstances studied.16 Thus estrogen, certainly in combination with progesterone, does not provide a panacea for protection against cardiovascular events. Decreased in high-density lipoprotein (HDL) levels in blood after menopause might explain part of the apparent premenopausal protection from cardiovascular risk.
High-density lipoprotein
Numerous animal studies have established the antiatherogenic effects of HDL or its major apolipoprotein (apo) AI, as have small biomarker studies in humans. Mechanisms by which HDL may protect against atherosclerosis include promotion of reverse lipid transport.17,18 Furthermore, nascent HDL particles can take up cholesterol from macrophages and other cells in a process that depends on the adenosine triphosphate (ATP) binding cassette transporter 1 (ABCA1). Mature HDL particles can also take up cholesterol through the related transporter ABCG1.19 In addition, HDL can have antiinflammatory effects.20 Mechanisms of HDL’s arterial protective effect may relate to its ability to bind and carry numerous proteins that may regulate lipid metabolism, oxidative stress, inflammation, and proteolysis.21
High-density lipoprotein’s metabolism is quite complex—more so in many respects than LDL.22 The steady-state level of HDL cholesterol, therefore, does not accurately predict cardiovascular protection. Although baseline levels of HDL cholesterol quite consistently track inversely with cardiovascular events, interventions that raise HDL cholesterol levels do not necessarily reduce event rates. Moreover, recent data challenge the extent to which HDL levels influence outcomes on individuals taking potent LDL-lowering treatment.23,24 The ability of HDL particles to effect cholesterol efflux from macrophages in vitro correlates with CAD status and the biomarker of risk, carotid intima media thickness.18 Yet the ability of pharmacologically induced increases in HDL to confer clinical benefit remains unproven. Several large-scale trials currently in progress should clarify whether drugs of several classes that elevate HDL can reduce clinical events.25,26
Emerging Risk Factors for Atherosclerosis
Homocysteine
Homocysteine, a product of amino acid metabolism, may contribute to atherothrombosis.27 Individuals with genetic defects that lead to elevated homocysteine levels (e.g., homocystinuria, commonly due to deficiency in cystathionine β-synthase) have a thrombotic diathesis. In vitro, treatment with homocysteine and related compounds can alter aspects of vascular cell function related to atherogenesis. Reliable clinical tests for hyperhomocysteinemia exist. Although clearly associated with elevated thrombotic risk in patients with homocystinuria, elevated levels of homocysteine in unselected populations only weakly predict cardiovascular risk (Fig. 8-1). Moreover, randomized trials that have used vitamin treatments to lower homocysteine levels have not documented improvements in cardiovascular outcomes.28–30 Enrichment of cereals and flour with folate in the United States has shifted dietary intake and should lower blood homocysteine levels in the American population.
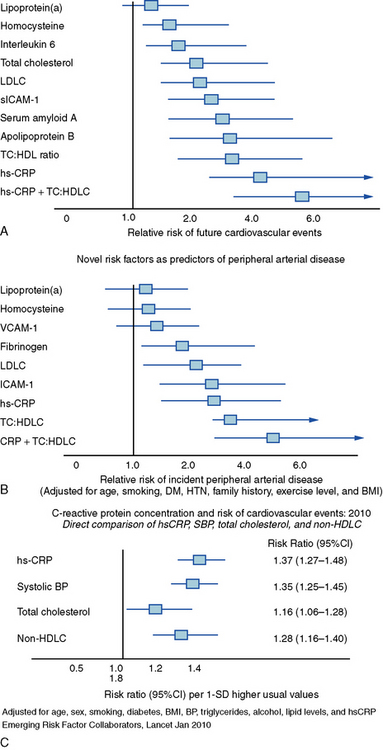
Lipoprotein(a)
Lipoprotein(a) (Lp[a]—commonly pronounced “L P little A”) consists of a low-density lipoprotein particle with apolipoprotein a (apo a) covalently attached to apo B, the major apolipoprotein of LDL particles. Apo a should not be confused with apo A, the family that includes apolipoprotein A-I, the principal apolipoprotein of HDL. Lipoprotein(a) has considerable heterogeneity, determined genetically and related to the number of repeats of a structural motif known as a kringle in the apo a moiety of the special lipoprotein particle. The structural resemblance of apo a to plasminogen suggests that Lp(a) may inhibit fibrinolysis. Lipoprotein(a) levels in the general population have high skew. Most individuals lie in the lower range of distribution, with a few “outliers” in the higher levels of Lp(a). Black Americans have a higher frequency of elevated Lp(a). Those with Lp(a) levels substantially above normal appear to have increased cardiovascular risk. As in the case of homocysteine, Lp(a) in large populations only weakly predicts future cardiovascular disease,31 but genetic studies suggest a causal role of Lp(a) in provoking cardiovascular events32 (see Fig. 8-1).
Fibrinogen
Fibrinogen, the substrate of thrombin, provides the major meshwork of arterial thrombi. Levels of fibrinogen increase in inflammatory states as part of the acute-phase response. A consistent body of observational evidence links elevated levels of fibrinogen with cardiovascular risk.33,34 Standardization of assays for fibrinogen has proved difficult. Moreover, diurnal variation in plasma fibrinogen levels weakens its potential as a biomarker of cardiovascular risk, despite its obvious biological plausibility as a major participant in thrombosis. Fibrin deposition in plaques, first hypothesized by von Rokitansky in the mid-19th century, provides evidence of fibrinogen’s involvement in atherogenesis.35
Infection
The possibility that infectious agents or responses to infection may contribute to atherogenesis or precipitate atherosclerotic events has periodically captured the fancy of students of atherosclerosis. Many infectious agents could contribute to aspects of atherogenesis by direct cytopathic effect or through mediators they release or elicit as part of a host defense.36–41 The hemodynamic stresses of acute infection, and accentuated thrombotic risk or impaired fibrinolysis due to acute-phase reactants such as fibrinogen or plasminogen activator inhibitor (PAI)-1, might transiently heighten the risk for thrombotic complications of atherosclerosis.42 Some seroepidemiological studies have suggested links between exposure to viral and bacterial pathogens and various measures of atherosclerosis or risk of atherosclerotic events. Prospective studies properly controlled for confounding factors have shown weak, if any, correlation of antibody titers against various microbial or viral pathogens and cardiovascular events. Trials of various antibiotic regimens in patients with CAD have not shown reductions in recurrent events.43
Inflammation and atherosclerosis
Recognition that inflammation provides a unifying theme for many of the pathophysiological alterations that occur during atherogenesis has provoked both interest and controversy.44–46 A subsequent section of this chapter will discuss in detail the links between inflammatory processes and risk factors, and atherogenesis and complications of atherosclerosis. Emergence of C-reactive protein (CRP) as a validated marker of prospective cardiovascular risk has spawned countless studies proposing other inflammatory markers as potential predictors of atherosclerotic risk.47–49 Although some markers of inflammation (e.g., fibrinogen, PAI-1) have defined roles as mediators and markers, evidence regarding CRP as an effector rather than a marker remains unsettled. Despite some laboratory evidence that CRP can produce some effects that may promote atherothrombosis, genetic data do not support a causal role for CRP in cardiovascular events.50–52 Of course, biomarkers need not participate directly in pathogenic pathways to have utility as gauges of risk, as illustrated by the case of CRP.53–55
Genetic predisposition
The example of familial hypercholesterolemia recounted earlier irrefutably illustrates the link between gene mutation and atherosclerosis. The accelerated development of molecular genetic technology and the increasing ease of identifying and cataloging genetic polymorphisms have facilitated the search for genetic variants that predispose toward atherosclerosis or its complications.56 Monogenic conditions such as familial hypercholesterolemia do not appear to explain the majority of the burden or risk of atherosclerotic disease. The quest for genetic polymorphisms that predispose toward atherosclerosis has yielded many potential candidates. Genome-wide association studies have identified reproducible regions of the genome associated with increased cardiovascular risk.57–61 Some of the genes at locations so identified have well-established functions in pathogenic pathways for atherosclerosis.62,63 Other sites emerging from genome-wide association studies have unknown functions and have not been previously associated with cardiovascular disease. Notably, the chromosome 9p21 region concordantly associates with cardiovascular events in several independent large population studies. Yet even combinations of well-validated genetic markers may fail to improve estimation of cardiovascular risk beyond a simple query regarding family history of premature CAD (at < 60 years of age).64,65 Genetic markers of atherosclerotic risk should nonetheless prove useful in providing new understanding of disease mechanisms and may identify new avenues for intervention, even if they do not add substantially to risk stratification.
Risk Factors on the Rise
We are witnessing a transition in the pattern of atherosclerotic risk factors in the United States, and indeed worldwide.66 Certain traditional atherosclerotic risk factors are on the wane. For example, rates of smoking in the United States are declining, particularly in men. Dissemination of effective antihypertensive therapies has provided a means to reduce the degree or prevalence of this traditional atherosclerotic risk factor. Although many patients do not achieve the currently established targets for blood pressure, effective therapies have become much more widely implemented in recent decades.
Although we justly derive considerable satisfaction from these pharmacological inroads into the traditional profile of cardiovascular risk, we are rapidly losing ground in other respects.66,67 The astounding increase in obesity in the U.S. population in the last decade alone represents a change in body habitus of substantial proportion in an instant on an evolutionary timescale. Short of major disasters or famines, this kind of rapid shift in body habitus may have no precedent in the history of our species. From the perspective of cardiovascular risk, the metabolic alterations that accompany this increased girth of our population should sound an alarm. Current data point to a significant increase in the prevalence of the components of the clustered risk factors often referred to as the metabolic syndrome.
The National Cholesterol Education Project Adult Treatment Panel III (ATP III) arbitrarily defined five components of the metabolic syndrome68 (Table 8-1); individuals with any three components have the syndrome, according to the ATP III criteria. There is disparity in the definitions of the metabolic syndrome among varied constituencies.69 Some have voiced cogent objections to the concept; for example, it is not clear that the sum of the risk of the metabolic syndrome components exceeds that of the individual factors combined, or that the components comprise a true syndrome, being linked mechanistically.70,71 But many clinicians find the construct practically useful insofar as it reflects the type of individuals seen more and more in practice.
Table 8-1 Criteria for the “Metabolic Syndrome”*
| Risk Factor | Defining Level |
|---|---|
| Abdominal Obesity† (Waist Circumference‡) | |
| Men | > 102 cm (> 40 in) |
| Women | > 88 cm (> 35 in) |
| Triglycerides | ≥ 150 mg/dL |
| HDLC | |
| Men | < 40 mg/dL |
| Women | < 50 mg/dL |
| Blood pressure | ≥ 130/≥85 mm Hg |
| Fasting glucose | ≥ 110 mg/dL |
HDLC, high-density lipoprotein cholesterol.
* Diagnosis is established when ≥ 3 of these risk factors are present.
† Abdominal obesity is more highly correlated with metabolic risk factors than is increased body mass index (BMI).
‡ Some men develop metabolic risk factors when circumference is only marginally increased.
From the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. JAMA 285:2486, 2001.
Note that LDL, the traditional focus of guidelines and therapies, does not figure among the metabolic syndrome criteria; instead, lower ranges of HDL and higher levels of triglycerides characterize the syndrome. Thus, we may witness a shift in lipid risk factor burden from primarily LDL to the dyslipidemia associated with obesity and/or insulin resistance, characterized by lower HDL and higher triglycerides. The success of statins in reducing LDL cholesterol will likely contribute to the increased importance of such dyslipidemia in the future. Although individuals with the metabolic syndrome cluster commonly have dyslipidemia, their levels of LDL cholesterol may be average or even below average. This finding may provide false reassurance to physicians and patients alike. Although LDL cholesterol levels in such patients may not be especially elevated, the quality of the lipoprotein particles may prove particularly atherogenic.72 Low-density lipoprotein particles in those with high levels of triglycerides and low levels of HDL tend to be small and dense. Such small, dense LDL particles appear to bind to proteoglycans in the arterial intima with more avidity. Their retention in the intima may facilitate their oxidative modification, so small, dense LDL particles may have particularly atherogenic properties.73 Prolonged retention and increased entry may promote lipoprotein accumulation in the artery wall.74
Hyperglycemia
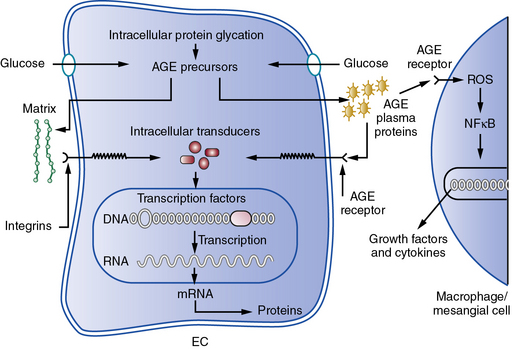
Hyperglycemia may contribute independently to the pathogenesis of atherosclerosis.75 In the presence of higher levels of glucose, nonenzymatic glycation and other types of oxidative posttranslational modification of various macromolecules increases. Hemoglobin A1c, a glycated form of this oxygen-carrying pigment, reflects this biochemical process. Accumulation of glycated macromolecules ultimately leads to the formation of complex condensates known as advanced glycation end products (AGEs) that may trigger inflammatory and oxidative responses implicated in atherogenesis.76 A cell surface receptor for AGEs known as RAGE may transduce proinflammatory signals when occupied by AGE-modified ligands77 (Fig. 8-2). This and other mechanisms link hyperglycemia and insulin resistance to aspects of host defenses considered essential for the atherogenic process, but strict glycemic control does not necessarily improve cardiovascular outcomes, as shown in several recent large clinical trials.78–81
Interactions of risk factors with cells in the arterial wall and leukocytes during atherogenesis
We increasingly understand atherosclerosis as a process that involves cellular interactions with risk factors such as high levels of LDL. This contemporary view contrasts with previous notions that the arterial wall passively accumulated cholesterol. This crosstalk among cells of varying types during atherogenesis involves more than just the intrinsic cells of the arterial wall, endothelium, and vascular smooth muscle cells (VSMCs) (see Chapters 2 and 3).44,82 Indeed, the mononuclear phagocyte also contributes importantly to atherogenesis. Normal endothelium resists prolonged contact with leukocytes, including blood monocytes, precursors of the tissue macrophages that accumulate in atheromata. A mechanism involving expression of particular leukocyte adhesion molecules on the endothelial surface likely mediates recruitment of blood monocytes to sites of formation of the earliest atherosclerotic lesions.83
The heterogeneity of monocytes and the macrophages to which they give rise has generated considerable recent interest.84–87 A particularly proinflammatory subset of monocytes accumulate in the blood of hypercholesterolemic mice. Macrophages exhibiting atherogenic functions also appear to accumulate in atherosclerotic lesions, and therapeutic interventions may modulate these functions.88
Once firmly bound to the endothelial surface, white blood cells must receive chemoattractant stimuli to penetrate into the intima. Among such signals, monocyte chemoattractant protein-1 (MCP-1) may be particularly important. Again, experiments in genetically altered mice support the involvement of MCP-1 in the formation of experimental atherosclerotic lesions. Other chemokines, such as the cell surface–associated molecule fractalkine, also may contribute to this process. In addition to mononuclear phagocytes, T lymphocytes accumulate in human atherosclerotic plaques, where they may play important regulatory roles. Adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1), overexpressed by ECs overlying atheromata, may participate with VCAM-1 in the adhesion of T lymphocytes. Moreover a trio of chemokines induced by the T-cell activator interferon (IFN)-γ may promote chemoattraction of adherent T cells into the arterial intima.89 Mast cells, long recognized in the leukocyte population of the arterial adventitia, also localize within the intimal lesions of atherosclerosis. Although vastly outnumbered by macrophages, mast cells may also contribute to lesion formation or complication.82,90–92 The chemokine exotaxin may participate in recruiting mast cells to the arterial intima.
Once present in the arterial intima, these various classes of leukocytes undergo diverse activation reactions that may potentiate atherogenesis. For example, monocytes mature into macrophages in the atherosclerotic plaque, where they overexpress a series of scavenger receptors that can capture modified lipoproteins that accumulate in the atherosclerotic intima. Because their levels do not decrease as cells accumulate cholesterol, these scavenger receptors permit formation of foam cells, a hallmark of the atheromatous plaque. Macrophages within the atherosclerotic intima proliferate and become a rich source of mediators, including reactive oxygen species and proinflammatory cytokines, that may contribute to progression of atherosclerosis.93
The T cells in the atherosclerotic plaque also appear to modulate aspects of atherogenesis.45,94 Interferon-γ, a strong activating stimulus for macrophages produced by activated type 1 helper T (TH1) cells, localizes in plaques. Indicators of the action of IFN-γ, such as induction of the class II major histocompatibility antigen molecules, provide evidence for the biological activity of IFN-γ in atherosclerotic plaques.
Once recruited to the intima, white blood cells can perpetuate, amplify, or mollify the ongoing inflammatory response that led to their recruitment. The function of the “professional phagocytes” adds to the proinflammatory mediators elaborated by the intrinsic vascular wall cells, and perpetuates and amplifies the local inflammatory response. T lymphocyte subsets may also quell inflammation. T helper 2 cells that produce interleukin (IL)-10, a putative antiatherosclerotic cytokine, also localize in plaques. Regulatory T cells (Treg) produce transforming growth factor (TGF)-β, another antiinflammatory and fibrogenic mediator that may modulate plaque biology.95 The role in atherogenesis of another T-cell subset, TH17, remains unsettled.96–98 Dendritic cells specialized in surveying the environment and presenting antigens to T cells arrive early in the arterial intima of mice subjected to hyperlipidemia.99–101 The nature of the antigenic stimulus to T-cell activation remains speculative, although animal experiments have suggested some candidates.102
Atherosclerosis progression
Recruitment of blood leukocytes and their activation in the arterial intima sets the stage for progression of atherosclerosis. Proinflammatory mediators produced by these various cell types lead to elaboration of factors that can stimulate migration of smooth muscle cells (SMCs) from the tunica media into the intima.103 The normal human tunica intima contains resident SMCs. Growth factors produced locally by activated leukocytes provide a paracrine stimulus to SMC proliferation. Activated SMCs also appear capable of producing growth factors that can stimulate their own proliferation or that of their neighbors, an autocrine pathway of proliferation.104 Smooth muscle cells also die in atheromata.105,106 Depletion of SMCs may influence the biology of plaques by disturbing repair and maintenance of the fibrous cap.107,108
Other mediators present in atheromatous plaques, such as TGF-β, can augment production of macromolecules of the ECM, including interstitial collagen.109,110 Thus, “maturing” atherosclerotic lesions assume fibrous as well as fatty characteristics. Ultimately, the established atherosclerotic plaque develops a central lipid core encapsulated in fibrous ECM. In particular, the fibrous cap—the layer of connective tissue overlying the lipid core and separating it from the arterial lumen—forms during this phase of lesion progression.
Calcification, another characteristic of the advancing atherosclerotic plaque, also involves tightly regulated biological functions. The expression of certain calcium-binding proteins within the plaque may sequester calcium hydroxyapatite. Such deposits, far from fixed, can undergo resorption as well as deposition. Inflammatory pathways participate in regulating mineralization of atheromata.111 Reminiscent of bone metabolism, activated macrophages within the plaque appear to function as osteoclasts. Indeed, mice deficient in M-CSF, the macrophage activator, show increased accumulation of calcified deposits.112 This observation supports the dynamic nature of calcium accretion in the plaque.
The atheroma eventually develops a central region filled with lipid, inflammatory cells, and cellular debris. Macrophage death, including apoptotic (programmed) death, contributes to formation of this lipid-rich core.105,113 Indeed, classical pathologists often referred to this region of the plaque as the necrotic core of atheroma. Defective clearance of apoptotic cells, a process denoted efferocytosis, may contribute to the formation of the plaque’s lipid core.114
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


