CHAPTER 7
Assessment of Stage C Patients with HF-pEF
“In the heart, the velocity and extent of relaxation, in other words, the ease with which the muscle stretches under the distending force of venous pressure is probably quite as important a factor in the heart’s behavior as the force and rapidity of the systolic contraction.”
–Yandell Henderson, 19231
Diagnosis of HF-pEF
Heart failure with preserved ejection fraction (HF-pEF) can be diagnosed when clinical findings of congestion due to elevated pulmonary or systemic venous pressures present with no more than mild left ventricular systolic dysfunction (EF > 40%). Although HF-pEF is associated with echocardiography findings of left ventricular diastolic dysfunction, this is not always the case (see Chapter 2). Similarly, left ventricular hypertrophy by echocardiography is often present, but HF-pEF may still be present due to coronary artery disease or other conditions without increased left ventricular wall thickness (Figure 7.1). Beyond pathologic processes that directly affect myocardial structure, inflammation from noncardiac comorbidities and increased arterial stiffness can indirectly contribute to cardiomyocyte hypertrophy, interstitial fibrosis, and impaired left ventricular diastolic filling.2
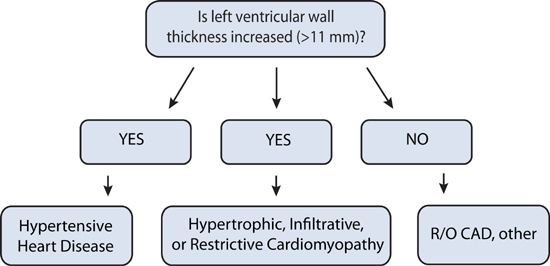
FIGURE 7.1 Causes of diastolic dysfunction and associated left ventricular wall thickness.
DIFFERENTIAL DIAGNOSIS
Infiltrates on chest x-ray and preserved systolic function by echocardiogram can also be seen with interstitial lung disease or non-cardiogenic pulmonary edema. However, interstitial lung disease does not improve with diuretics. Non-cardiogenic pulmonary edema (also called adult respiratory distress syndrome) can occur in a patient with an acute severe noncardiac systemic illness. In this setting, lung edema develops due to a “capillary leak” despite normal left heart filling pressures. BNP may be normal or mildly elevated secondary to right ventricular strain.
In most patients, clinical findings and Doppler echocardiographic assessment (see List 7.1) are adequate to distinguish these conditions from HF-pEF. In some cases with indeterminate or overlapping findings, right heart catheterization should be performed; pulmonary capillary wedge pressure is high in HF-pEF and normal or low in primary pulmonary disorders.
LIST 7.1 Echo-Doppler Findings in Diastolic Dysfunction
ECHOCARDIOGRAPHIC FINDINGS WITH DIASTOLIC DYSFUNCTION
Diastolic filling of the left ventricle can be assessed by Doppler echocardiography.3 Left atrial enlargement can indicate the presence of longstanding structural heart disease.
Echocardiographic measurement of left atrial size by dimension or calculated volume has been called the “hemoglobin A1C” of left atrial pressure and, when increased, serve as an index of chronically elevated left-sided heart filling pressures.4 In patients with symptomatic HF-pEF, progressive shortening of the transmitral deceleration time (DT) and increasing E/A ratio can be seen with decreasing ventricular compliance and increasing left atrial pressure (Figure 7.2).5 Acute alterations in mitral inflow velocities may occur in response to patient treatment or other changes in hemodynamic status.
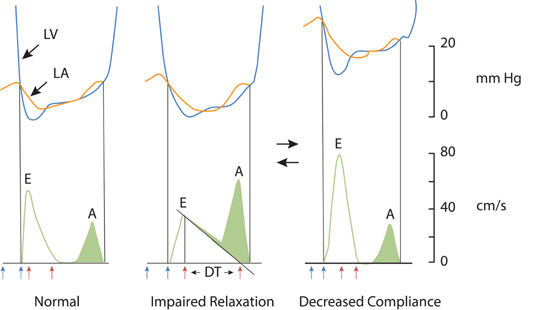
FIGURE 7.2 Doppler echocardiographic assessment of left ventricular diastolic filling. Changes in Doppler mitral velocities with correlation to left ventricle (blue) and left atrial (orange) pressures during diastole. Filling velocities and extrapolated deceleration times (red arrows) are in response to the transmitral pressure gradient. Blue arrows indicate interval of isovolumic relaxation time from aortic valve closure to mitral valve opening. Abbreviations: LV, left ventricle; LA, left atrium; E, early diastolic mitral inflow; A, diastolic filling during atrial systole; DT, deceleration time.5 Source: Adapted with permission from Nagueh et al., Eur J Echocardiogr. 2009;10(2):165-193.
Tissue Doppler imaging (TDI) of mitral annulus motion can also assess myocardial relaxation. With systolic ejection of blood, there is contraction of the left ventricle in part achieved by mitral annular descent toward a relatively fixed apex. Following this, during ventricular filling, the annulus returns towards its initial position (Figure 7.3). Tissue Doppler imaging (TDI) displays the velocity profile of these movements. The velocity of the mitral annulus away from the left ventricular apex during early diastole (e′) reflects the rate of myocardial relaxation and may be less dependent on pressure gradients than transmitral blood flow velocity.3
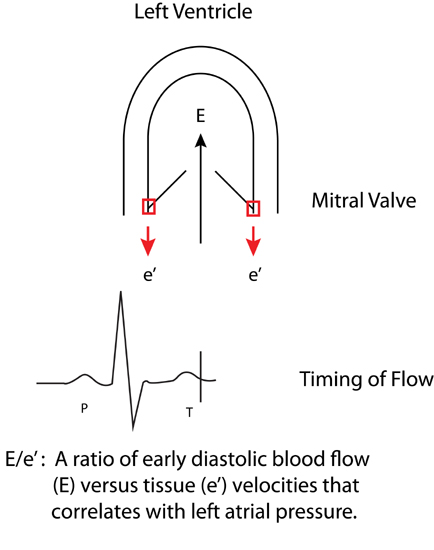
FIGURE 7.3 Derivation of the E/e ′ ratio. Transmitral early diastolic blood flow (E) and mitral annulus tissue velocities (e′). High left atrial filling pressures with impaired diastolic filling are associated with increased blood flow E and decreased e′ tissue velocities.
In HF-pEF, the E/e′ ratio can be used as an initial measurement for an estimate of left ventricular filling pressures (Figure 7.4). Because of its utility, Doppler echocardiography has been called the “Rosetta Stone” for evaluation of diastolic function.6
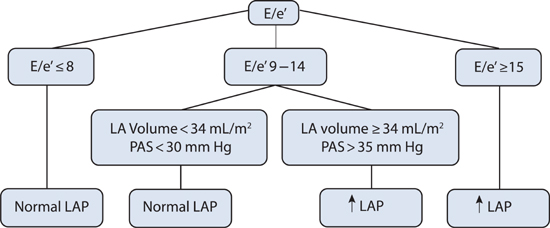
FIGURE 7.4 Diagnostic algorithm for estimating left ventricular filling pressures based on Doppler echocardiographic findings in patients with HF-pEF. Abbreviations: E, early diastolic transmitral blood flow; e′, mitral annulus tissue velocity; LA, left atrium; PAS, pulmonary artery systolic pressure; LAP, left atrial pressure.5 Source: Adapted with permission from Nagueh et al., Eur J Echocardiogr. 2009;10(2):165-193.
KEY DIAGNOSTIC FEATURES OF HYPERTENSIVE HEART DISEASE
When a patient with HF-pEF has a history of high blood pressure and uniform left ventricular hypertrophy by echocardiogram, the diagnosis of hypertensive heart disease is likely.
Echocardiographic findings of diastolic dysfunction support this diagnosis. Consider that patients with hypertensive heart disease may also have associated coronary artery disease (CAD). Ventricular hypertrophy in the absence of a history of high blood pressure or CAD may imply the presence of a secondary process due to hypertrophic, infiltrative, or restrictive cardiomyopathy (see descriptions below).
KEY FEATURES OF HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy (HCM) can be defined as left and/or right ventricular hypertrophy occurring usually in an asymmetric pattern and often involving the interventricular septum not secondary to systemic hypertension or other systemic disease.7 Left ventricular chamber volume is normal or reduced. Microscopically, there is myocyte hypertrophy and disarray surrounding areas of increased loose connective tissue. When hypertrophic cardiomyopathy is associated with an obstructive gradient across the left ventricular outflow tract (LVOT), either at rest or after provocation, management directed toward improving this gradient may be important. End-stage hypertrophic cardiomyopathy may progress to systolic dysfunction. Although many terms have been used historically to describe HCM, including idiopathic hypertrophic subaortic stenosis (IHSS) and hypertrophic obstructive cardiomyopathy (HOCM), currently, using the term hypertrophic cardiomyopathy (HCM) and additional descriptive features is preferred (List 7.2).
LIST 7.2 Phenotypes of Hypertrophic Cardiomyopathy
Within the spectrum of patients with heart failure, patients with hypertrophic cardiomyopathy represent a distinct subset because treatment options differ. Hypertrophic cardiomyopathy typically arises from either an inherited or spontaneous point mutation in genes coding for proteins within the sarcomere including the heavy chain of myosin (see Chapter 4). The prevalence of all forms of hypertrophic cardiomyopathy may be as common as 1 in 500 in the United States population; however, many patients are asymptomatic.8 The location of regional or global hypertrophy within the left (or right) ventricle between individuals can vary, even within a single family. Other functional features include diastolic dysfunction, mitral regurgitation, myocardial ischemia, and arrhythmias.
Echocardiography or cardiac magnetic resonance imaging (MRI) can be used to visualize the distribution of hypertrophy (Figure 7.5). The most common pattern is asymmetric septal hypertrophy with a ratio of septal to posterior wall thickness of 1.3 or greater. When there is dynamic outflow tract obstruction, a characteristic “spike and dome” morphology may be observed in the aortic pressure waveform or LVOT velocity. This pattern arises from an initial unobstructed ejection of blood from the left ventricle followed by progressive obstruction of outflow during the period of mid-to-late systolic ejection. An increase in systolic Doppler velocity across the LVOT narrowed by septal hypertrophy and systolic anterior motion (SAM) of the mitral valve (Figure 7.5) can be observed at rest or following physiologic provocation such as after premature ventricular contractions, post-exercise, or during the strain phase of the Valsalva maneuver (Figure 7.6). Approximately one-third of patients have nonobstructive HCM defined as resting or peak gradient after provocation of < 30 mm Hg. Patients with resting or provocable gradients ≥ 50 mm Hg and persistent symptoms may benefit from surgical or percutaneous intervention.7
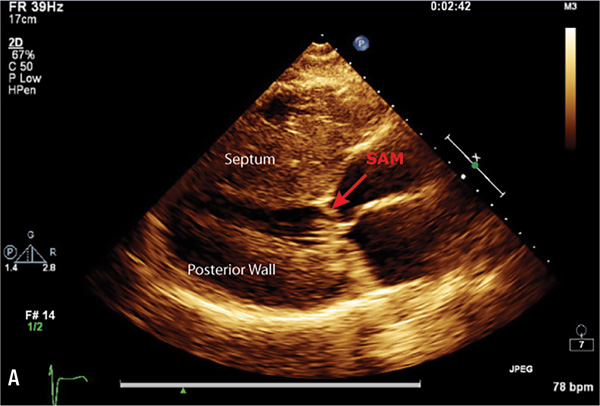
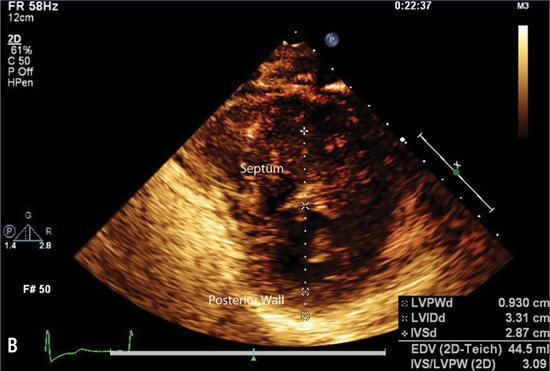
FIGURE 7.5 Imaging by 2D echocardiogram of cardiac abnormalities caused by hypertrophic cardiomyopathy. Images show a 28-year-old female with HCM. Parasternal long axis (Panel A) reveals Systolic Anterior Motion (SAM) of the mitral valve leaflets. Echo parasternal short axis (Panel B) demonstrates asymmetric septal hypertrophy (left ventricle end-diastolic thickness: Septum measurement = 2.9 cm, Posterior wall = 0.9 cm).
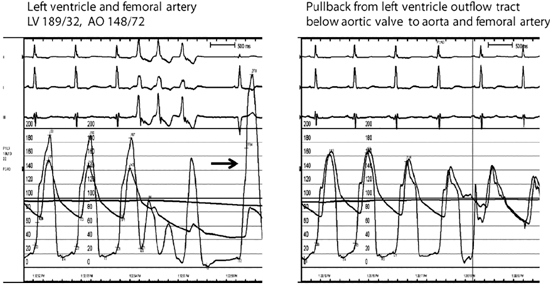
FIGURE 7.6 Hemodynamics and provocation maneuvers in HCM with dynamic LVOT obstruction. Left panel: Example of patient with a resting peak systolic left ventricular-aortic gradient of 41 mm Hg that increased to 192 mm Hg in a post-PVC beat (arrow). Despite the markedly increased left ventricular pressure of the post-PVC beat, arterial pulse pressure decreased (known as the Brockenbrough-Braunwald sign). Elevated left ventricular end-diastolic pressure of 32 mm Hg is consistent with diastolic dysfunction of the hypertrophic ventricle. Moderate to severe mitral regurgitation was also present. Right panel: No aortic valvular gradient was present during pullback from just below the aortic valve to the aorta, thus excluding aortic valve disease as contributing to the gradient. After surgical septal myectomy (data not shown), dynamic outflow tract gradient completely resolved.
Diffuse concentric hypertrophy of the left ventricle is another type of hypertrophic cardiomyopathy. However, this pattern of hypertrophy may also be seen with hypertensive, athletic, or infiltrative causes of hypertrophy. When global hypertrophy is detected and there is no history of hypertension or family history of HCM, additional diagnostic tests may be indicated. Cardiac MRI may identify delayed gadolinium hyperenhancement consistent with a HCM pattern of fibrosis (see Figure 6.16) or, endomyocardial biopsy may be needed when infiltrative causes are suspected.9
A less common manifestation of hypertrophic cardiomyopathy is hypertrophy confined to the apex of the left ventricle. This pattern often displays marked T-wave inversion across the precordial leads on a standard 12-lead electrocardiogram.
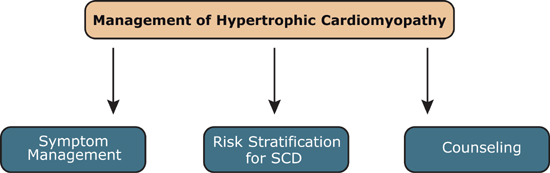
Management of Hypertrophic Cardiomyopathy
Management of hypertrophic cardiomyopathy (HCM) includes three components: symptom management, risk stratification for sudden cardiac death (SCD), and counseling.10
SYMPTOM MANAGEMENT
Two common symptoms of HCM are exertional dyspnea and chest pain. Chest pain often is not due to epicardial artery stenosis, but rather to functional ischemia due to increased myocardial oxygen demand from hypertrophy exceeding limited endocardial supply. Beta-blockers that decrease contractility and heart rate can lead to hemodynamic improvement in HCM by decreasing outflow tract obstruction and functional myocardial ischemia. The calcium channel blocker verapamil has negative inotropic and bradycardic effects that may also improve left ventricular outflow obstruction. However, it should be used cautiously, because its action as an arteriolar vasodilator may increase the dynamic outflow tract gradient. If these medications are poorly tolerated, the antiarrhythmic-negative inotropic agent disopyramide may be considered as an alternative.
In HCM with dynamic outflow tract obstruction, medications that increase myocardial contractility, such as digoxin or catecholamines, should be avoided. Also, vasodilators or diuretics should be used cautiously because they can reduce left ventricular size and worsen left ventricular outflow obstruction and gradient.
In patients with persistent, symptomatic HCM and obstructive physiology, invasive therapies may be appropriate, including surgical myectomy or septal ablation using percutaneous catheter infusion of alcohol.11 Previously, dual-chamber (atrial-ventricular) pacing with right ventricular electrical activation was considered for palliation in patients who were high risk for surgery.12 This has largely been superseded by septal alcohol ablation.
Paroxysmal, persistent, or permanent atrial fibrillation can exacerbate symptoms in HCM. Electrical cardioversion may be needed to rapidly restore sinus rhythm. To maintain sinus rhythm, disopyramide, sotalol, or amiodarone may be used. Catheter ablation or surgical Maze procedure for prevention of recurrent atrial fibrillation may be required in persistent cases.13
SUDDEN CARDIAC DEATH IN HCM
Patients with HCM may have an increased risk for Sudden Cardiac Death (SCD) due to ventricular tachycardia or fibrillation. In high-risk individuals, implantable cardioverter-defibrillators (ICDs) can be more effective compared to drugs alone such as beta-blockers and amiodarone.7 Identification of risk factors for SCD can help guide appropriate recommendations for ICD implant (List 7.3).
LIST 7.3 Risk Factors for SCD in HCM
One point for each factor: • Family history of sudden death • Unexplained syncope • Nonsustained ventricular tachycardia on ambulatory monitoring (3 or more beats ≥ 120 bpm) • Abnormal hypotensive blood pressure response (< 20 mm Hg increase or drop ≥ 20 mm Hg during exercise) to treadmill exercise testing (in patients < 50 years old) • Severe left ventricular hypertrophy (> 30 mm) | |
Risk Factors Score | Recommendation |
• 0 | • Reassurance |
• 1 | • Individualize |
• 2+ | • Recommend ICD |
• Prior SCD | • Recommend ICD |
• Sustained VT | • Recommend ICD |
|
|
Cardiac MRI with late gadolinium enhancement can provide additional assessment of myocardial pathology. It has been proposed that visualization of myocardial scar in the area of left ventricular hypertrophy by this technique can be used to support decision making regarding recommendations for ICD implantation.14 At present, the decision making for ICD implantation is based upon age, number and nature of risk factors, and clinical judgment.10
GENETIC VARIANTS OF HCM
In individuals with HCM, genetic mutations associated with hypertrophic cardiomyopathy may be identified in approximately 60% to 70% of those with a positive family history, but only 10% to 50% of those without a family history (see Chapter 4).7 Genetic testing from a blood sample may be considered when identification of a known mutation may help with screening family members. A negative genetic test does not exclude the potential to develop hypertrophic cardiomyopathy, unless screening fails to find a specifically identified mutation matched to an affected family member.
Approximately 5% of families with HCM will have 2 or more sarcomere mutations15 that may be associated with a greater risk for sudden cardiac death.16
HCM with delayed penetrance and phenotypic expression may not be manifest until later in life. If an affected patient does not have a known mutation, then periodic imaging, usually by echocardiography, is used for phenotypic family screening of first-degree relatives.17
COUNSELING
Counseling is important in caring for the patient with HCM for several reasons. Many types of HCM have a benign prognosis and it may be important to emphasize that the annual mortality in asymptomatic patients without high risk SCD or genetic findings may be less than 1%.18 Asymptomatic individuals may prefer serial echo to gene testing to monitor risk for cardiomyopathy. Exercise guidelines are available, especially for individuals who are diagnosed with HCM at a young age.19 In general, these guidelines have to be individualized to the severity of HCM and the type of exercise. Exercise treadmill testing can help assess the functional status of a patient with HCM for specific activities.
Restrictive Cardiomyopathy Due to Amyloidosis
The most common identifiable cause of restrictive cardiomyopathy is amyloidosis. Four types of amyloidosis vary in prognosis and natural history (Table 7.1). One of the most severe is cardiac involvement from AL amyloidosis associated with immunoglobulin light chain deposition and plasma cell dyscrasia. Two different forms of amyloidosis may occur due to misfolding, aggregation, and deposition of transthyretin (a circulating protein produced by the liver that transports thyroxin and retinol). Familial amyloidosis (ATTR) is due to a mutation that increases this misfolding. Senile amyloidosis due to wild-type transthyretin protein can also lead to cardiac involvement, but is usually less aggressive and occurs late in life, predominantly in males. Amyloidosis secondary to chronic inflammation is not commonly associated with cardiac involvement.20
TABLE 7.1 Types of amyloidosis.20
PHENOTYPE (NOMENCLATURE): AMYLOID FIBRIL PRECURSOR | ORGAN INVOLVEMENT | TREATMENT |
LIGHT CHAIN (AL): Immunoglobulin light chain | Heart Other: kidney, liver, peripheral/autonomic nerves, soft tissue, gastrointestinal system | Chemotherapy |
FAMILIAL (ATTR): Mutant transthyretin (TTR) | Heart Peripheral/autonomic nerves | • Liver transplantation • New pharmacologic strategies to stabilize the TTR/tetramer (if cardiac involvement is present, cardiac amyloid may progress despite liver transplantation) |
SENILE SYSTEMIC AMYLOID: Wild-type transthyretin | Heart | Supportive |
INFLAMMATORY (AA): Serum amyloid A | Kidney Heart (rarely) | Treat underlying inflammatory process |
< div class='tao-gold-member'>



