Medical device recalls have called attention to the device approval process in the United States. The premarket approval (PMA) process requires clinical trials to evaluate safety and effectiveness, whereas the expedited 510(k) process does not. The 510(k) process has been considered a source of increased recalls. This study aimed to assess the relative safety of medical device approval pathways based on the numbers of approvals and recalls. Data on recalls in the United States from January 2005 to December 2012 were collected from the Food and Drug Administration Web site. Over 8 years, 30,002 devices were approved, 5,728 by PMA (19%) and 24,274 (81%) by 510(k). There were 249 recalls due to serious risks, 0.45% of PMA approvals, and 0.92% of 510(k)-cleared devices, p <0.001. Over 1/2 of the recalls were during the first 2 years on the market. Percentage of recalled PMA devices was unchanged over the 8 years, whereas 510(k) recalls increased in 2010 to 2012 (from 0.65% to 1.39%, p <0.001). Cardiovascular devices represent the largest class of recalls (27%). The proportions of recalled PMA and 510(k) cardiovascular devices were the same as for all medical devices until 2011, but 510(k) recalls dramatically decreased in 2012 to the lowest recall rate seen (0.73%). In conclusion, recall rates were the same for 510(k)- and PMA-approved devices in 2005 to 2009 and increased for 510(k) devices subsequently. Modifying the 510(k) process with more rigorous performance testing, a conditional 2-year approval and a mandatory registry may be an approach to reduce recalls.
Medical devices play a critical role in health care in the United States with millions of patients depending on them for the diagnosis and management of disease. However, medical devices can cause adverse events. The Federal Food, Drug, and Cosmetic Act requires a reasonable assurance of safety and effectiveness before a device can be marketed. The framework that the Food and Drug Administration (FDA) regulates the device market is based on the “Medical Device Amendments” passed by Congress in 1976. Briefly, the FDA recognizes 3 classes of medical devices. The classification is based on the device complexity, technical characteristics, degree of invasiveness, and potential for harm if misuse or malfunction occurs.
Class I medical devices are low-risk devices that are not intended for supporting life or prevention of impairment to human health, and most are exempt from the “Premarket Notification” that is also called 510(k) review pathway. Class II devices (powered wheelchairs, infusion pumps, surgical drapes, and so on) are considered moderate risk with most cleared by 510(k) pathway. If the manufacturer can show that its device is substantially equivalent to a predicate, additional clinical data are usually not required. The chain of previous predicates can go back in time to before 1976.
Class III devices are high-risk devices, usually those that support or sustain life, are of substantial importance in preventing impairment, or present a potential of unreasonable risk of illness or injury if misused or malfunction would occur. Examples of Class III devices include implantable pacemakers, pulse generators, human immunodeficiency virus diagnostic tests, external defibrillators, and orthopedic implants. Class III devices require premarket approval (PMA) based on clinical trials. However, a number of Class III medical devices are cleared by the 510(k) process, if the device was found substantially equivalent to a device that previously was cleared through the 510(k) pathway.
Some high profile cases of medical device recalls have attracted attention. A number of reports have been critical of the 510(k) approval process. One study reported that most medical devices recalled for life-threatening or very serious hazards were originally cleared for market using the 510(k) process. The authors of these reports advocate reform of the regulatory process. However, the larger number of recalls in the 510(k) group could be related to the much larger number of devices approved by this pathway.
The FDA turned to the Institute of Medicine (IOM) to review the 510(k) process. The IOM report stated that the 510(k) process is “fundamentally flawed,” lacks the legal basis to be a reliable premarket screen for the safety and effectiveness of moderate-risk devices, that it cannot be transformed into a well-performing process, and therefore should be replaced with a completely different system. The FDA did not accept the conclusions of the IOM report, although stating that the process did need improvement. The device industry rejected the findings of the IOM report with some defending the status quo, whereas others called for revising the 510(k) process rather than scrapping it.
Several reports have tried to evaluate the effectiveness and safety of 510(k) approval process, although by not using a quantitative approach. The present analysis aimed to compare the 510(k) and PMA approvals and recalls on the basis of the number of devices approved in each group.
Methods
Data on all medical devices approved and recalled in the United States from January 2005 to December 2012 were collected from the FDA Web site ( www.FDA.gov ). Monthly listings of Premarket Notifications, that is, 510(k), and PMAs were identified to calculate the total number of medical devices approved by PMA or cleared by 510(k) process for each year. Data on medical device recalls were obtained from the FDA’s “Medical Device Recalls” Web site. At this Web site, the FDA posts information about the most serious medical device recalls listing only those devices that posses “a reasonable chance that they could cause serious health problems or death.” The time on the market was calculated using the time between the first and last manufacturing and/or distribution dates or the time between approval and recall. Additional information on recalled devices was retrieved from the FDA Medical and Radiation-Emitting Recalls search engine and from the FDA Manufacturer and User Facility Device Experience search engine. The collected data included date of approval, device class, approval process (PMA, 510(k), or exempt), and device classification. Using the total number of approvals and recalls, the proportions of recalls for PMA and 510(k) groups were calculated. The data were analyzed by Fisher’s exact test. The time on market before recall was calculated in a similar way and analyzed by independent sample t test. A 2-sided test with p <0.05 was considered to indicate a significant difference.
Results
From January 2005 to December 2012, a total of 30,002 medical devices were approved in the United States. A total of 5,728 were approved by PMA (19%) and 24,274 were cleared by the 510(k) process (81%). Table 1 lists the number of yearly approvals by PMA or 510(k). Whereas PMA increased steadily from 465 in 2005 to 894 in 2012, the number of devices that were marketed by 510(k) clearance remained steady. Despite an increase in the number of PMA-approved devices, they remained a relatively small portion of approvals.
| Year | Cleared by 510(k) | Approved by PMA | Recalled 510(k) | Recalled PMA | Recalled % of 510(k) | Recalled % of PMA | p Value |
|---|---|---|---|---|---|---|---|
| 2005 | 3,148 | 465 | 20 | 3 | 0.54 | 0.65 | 0.980 |
| 2006 | 3,200 | 662 | 17 | 1 | 0.53 | 0.15 | 0.342 |
| 2007 | 2,975 | 658 | 20 | 4 | 0.64 | 0.61 | 0.789 |
| 2008 | 3,031 | 701 | 14 | 4 | 0.40 | 0.57 | 0.761 |
| 2009 | 2,991 | 680 | 28 | 3 | 0.84 | 0.44 | 0.158 |
| 2010 | 2,766 | 708 | 43 | 2 | 1.48 | 0.28 | 0.005 |
| 2011 | 3,072 | 960 | 38 | 3 | 1.24 | 0.31 | 0.010 |
| 2012 | 3,091 | 894 | 43 | 6 | 1.39 | 0.67 | 0.006 |
| Overall | 24,274 | 5,728 | 223 | 26 | 0.92 | 0.45 | 0.0004 |
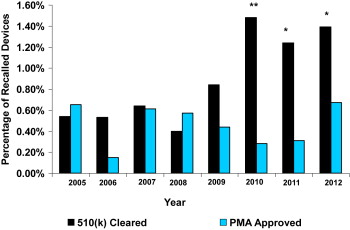
During the 8-year study period, a total of 249 recalls were identified in the Medical Device Recalls database. At this Web site, the FDA posts information about the most serious medical device recalls. Of the 249 recalls, 246 were Class I recalls (most serious) and 3 were Class II recalls. Class I recalls are defined as a situation in which there is a reasonable probability that the use of or exposure to a product will cause serious adverse health consequences or death. Of the 249 recalled devices, 26 were approved by PMA representing 0.45% of all PMA-approved devices and 223 were cleared by the 510(k) process representing 0.92% of all 510(k)-approved devices (p <0.001). Although the number of recalls of 510(k)-cleared devices were almost 9 times more frequent than the number of PMA-approved devices (223 vs 26), the absolute number of all 510(k)-cleared devices was also larger (4.3 times) resulting in a much smaller difference in the percentage of recalled devices ( Table 1 ). The percentage of recalled PMA-approved devices remained constant over the 8-year study period ( Figure 1 ). In contrast, the percentage of recalled 510(k)-cleared devices dramatically increased in 2010 and remained higher in 2011 and 2012 than in the first 5 years studied. From 2005 to 2009, the percentages of PMA and 510(k) recalled devices were similar. The percentage of recalls during these 5 years did not differ significantly (PMA 0.47%, 510(k) 0.65%, p = 0.32). The percentages of 510(k) recalled devices were significantly higher than the percentages of PMA recalled devices in 2010 (1.48% vs 0.28%, p <0.005), in 2011 (1.24% vs 0.31%, p <0.01), and in 2012 (1.39% vs 0.67%, p <0.01). When the number of recalls in 2010 to 2012 were combined, the difference between recalled 510(k)- and PMA-approved devices became more apparent (1.39% vs 0.43%, p <0.005). Of the 249 recalled devices, 52 were Class III devices, 26 received marketing approval by the PMA process (50%), and 26 were cleared by the 510(k) pathway (50%).
Recalls of cardiovascular devices represent the largest class of recalls, 27% of all recalls in the most serious database. From January 2005 to December 2012, a total of 5,437 cardiovascular devices were approved, representing approximately 1/5 (18%) of all device approvals. The proportions of PMA-approved cardiovascular devices were much greater than the proportions of PMA approvals for all medical devices. Almost 1/2 (46%) of the cardiovascular devices were approved by the PMA process ( Table 2 ), whereas PMA approvals were only 19% of all medical devices ( Table 1 .)
| Year | Cleared by 510(k) | Approved by PMA | Recalled 510(k) | Recalled PMA | Recalled % of 510(k) | Recalled % of PMA | p Value |
|---|---|---|---|---|---|---|---|
| 2005 | 339 | 243 | 6 | 3 | 1.77 | 1.23 | 0.742 |
| 2006 | 373 | 411 | 4 | 0 | 1.07 | 0 | 0.052 |
| 2007 | 317 | 294 | 4 | 3 | 1.26 | 1.02 | 0.890 |
| 2008 | 352 | 320 | 4 | 0 | 1.14 | 0 | 0.126 |
| 2009 | 394 | 279 | 9 | 2 | 2.28 | 0.72 | 0.110 |
| 2010 | 378 | 267 | 13 | 2 | 3.44 | 0.75 | <0.05 |
| 2011 | 390 | 304 | 11 | 1 | 2.82 | 0.33 | <0.02 |
| 2012 | 410 | 366 | 3 | 1 | 0.73 | 0.27 | 0.373 |
| Overall | 2,953 | 2,484 | 54 | 12 | 1.83 | 0.48 | <0.001 |
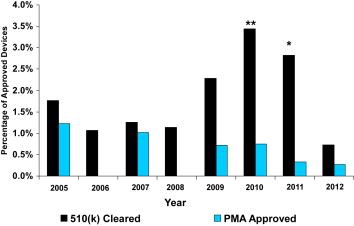
During the 8-year study period, a total of 66 cardiovascular device serious recalls were identified. Of those, 12 devices were approved by PMA, representing 0.48% of all PMA-approved cardiovascular devices, and 54 were cleared by the 510(k) process, representing 1.83% of all 510(k)-approved devices (p <0.001). The percentages of recalled devices for each type of approval for each year are listed in Table 2 . The trends of the percentage of recalled PMA and 510(k) devices are the same as seen for all recalled medical devices until 2011 as shown in Figure 2 . The percentage of recalled PMA-approved devices did not indicate any clear trend and, in fact, remained a small percentage of devices approved during the 8-year study period. In contrast, the percentage of recalled 510(k)-cleared cardiovascular devices dramatically increased in 2009, increased further to 3.4% in 2010, and remained high in 2011 (2.8%). The percentage of recalled 510(k)-cleared cardiovascular devices dramatically decreased in 2012 with the lowest recall rate (0.73%) seen. In 2010 and 2011, the percentages of 510(k) recalled cardiovascular devices were significantly higher in each year than the percentages of PMA recalled devices (3.44% vs 0.75%, p <0.05% and 2.82% vs 0.33%, p <0.02), whereas no significant differences were found in 2012 (510(k) 0.73% vs PMA 0.27%, p = 0.373).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


