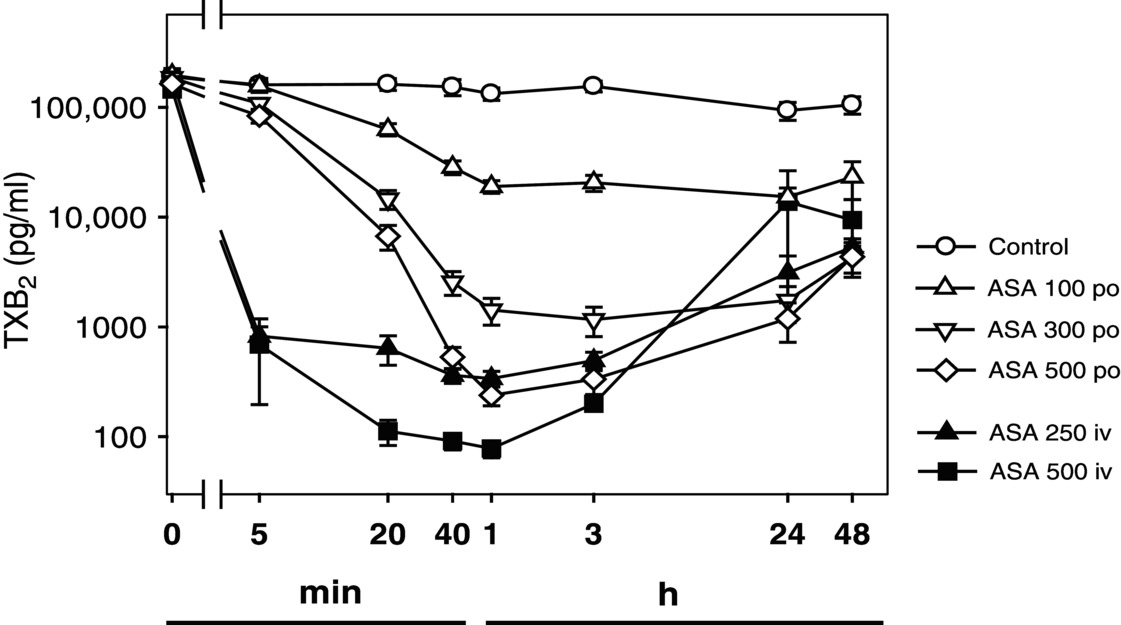
13 Karsten Schrör and Thomas Hohlfeld UniversitätsKlinikum, Heinrich-Heine Universität Düsseldorf, Düsseldorf, Germany Aspirin is the first-choice antiplatelet agent in cardiocoronary prevention and an essential constituent of dual antiplatelet therapy. The reasons are its unique pharmacological mode of action; the long-lasting, more than 60 years, experience with its use as an antiplatelet agent; and the reliability of its pharmacological action in more than 95% of patients. Aspirin, in contrast to most other antiplatelet drugs, is not a platelet-specific agent but exhibits a broad spectrum of biological activities on many cellular targets [1]. At low doses (≤300 mg), aspirin inhibits platelet-dependent prostaglandin-endoperoxide (PGEP) formation via irreversible acetylation of COX-1. This results in downstream inhibition of thromboxane A2 (TXA2) formation, the only significant PGEP end product in platelets. This COX-1-mediated inhibition of prostaglandin formation is the generally accepted mode of antiplatelet action of aspirin [2, 3]. Consequently, measurement of platelet-dependent thromboxane formation, for example, by determining thromboxane-forming capacity in serum ex vivo or in vitro, is a simple and specific means to determine the pharmacological potency of aspirin as an antiplatelet drug on an individual basis. Because of the nonlinearity between thromboxane levels and thromboxane-related platelet stimulation, this inhibition has to be apparently complete, that is, >95% in terms of serum capacity, to become effective. This degree of inhibition is obtained in the vast majority of patients, while only 1–2% remain pharmacologically “resistant” against aspirin. Thus, the much more variable clinical efficacy of aspirin in preventing atherothrombotic cardiovascular events is due to variable environmental conditions of circulating platelets including platelet stimuli, which might or might not require platelet-dependent thromboxane formation as an amplifying event. Accordingly, the antiplatelet potency of aspirin in vivo is critically determined by the significance of thromboxane generation for platelet activation. This largely depends on the stimulus. Aspirin does not modify ADP-induced platelet aggregation and alpha-granule secretion in native blood. Nor does it prevent platelet aggregation induced by thrombin, shear stress, or other thromboxane-independent pathways of platelet activation. The initial “explosion” of platelet-dependent thromboxane formation after appropriate stimuli starts platelet activation, secretion, and aggregate formation. However, the subcellular targets of thromboxane in platelets and their interactions are still not completely understood. At least theoretically, it is likely that thromboxane per se rather acts as a platelet-derived amplification factor for further platelet recruitment and activation than being a direct stimulus for platelet secretion [4, 5]. There is considerable clinical and experimental evidence for aspirin-sensitive, thromboxane-dependent thrombin formation, which occurs at the surface of activated platelets and connects platelet activation with the plasmatic clotting process [6, 7, 8, 9]. If rapid inhibition of platelet function is required, that is, as an emergency first-line treatment in acute coronary syndromes, a loading dose of 250–500 mg soluble aspirin, for example, as lysine salt, can be administered IV. This results in sufficient (≥99%) inhibition of thromboxane formation within 5 min. For oral dosing of 500 mg, at least 40 min is required for a comparable effect [10] (Figure 13.1). After oral administration, more time is necessary to obtain the required pharmacological effect. At least two to three daily doses (e.g., 75–325 mg) of standard aspirin are necessary to obtain sufficient inhibition of thromboxane formation. Once sufficient acetylation is obtained, only a maintenance dose of aspirin per day is necessary. Figure 13.1 Time-dependent inhibition of thromboxane (TXB2) formation by aspirin, depending on dose (mg) and route of administration. Data were obtained from 21 healthy subjects treated in a crossover design. Note that both axes are in logarithmic scale. (Source: Adapted from Schrör, K 2009 [1]. Reproduced with permission of John Wiley & Sons Ltd.) The inhibition of platelet COX-1 and platelet function by aspirin is functionally antagonized by the 10–15% fresh platelets that enter the circulation every day from the bone marrow. In certain clinical conditions with a more rapid platelet turnover rate, that is, diabetes or extracorporeal circulation, the efficacy and duration of the antiplatelet effect of aspirin might be reduced. A nearly complete recovery of (arachidonic acid-induced) platelet aggregation is seen about 3 days after aspirin withdrawal, but more than 10 days are required to fully restore plasma thromboxane levels [11]. This confirms that a small recovery, that is, 10–15%, even a small recovery of thromboxane formation is sufficient for a functionally significant thromboxane-induced platelet response.
Aspirin
Mode of antiplatelet action
Time dependency of inhibition of platelet function by aspirin
Dose-dependent inhibition of platelet function by aspirin
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


