Aspergillus, Candida, and Other Opportunistic Mold Infections of the Lung
PULMONARY ASPERGILLOSIS
Aspergillus is a ubiquitous saprophytic mold that plays an essential role in recycling carbon and nitrogen.1 This fungus has a simple biological cycle characterized by a high sporulating capacity, which results in the release of conidia at high concentrations (1–100 conidia/m3) into the atmosphere. Aspergillus conidia have a diameter small enough (2–3 μm) to reach the pulmonary alveoli.1,2 However, while humans constantly inhale Aspergillus conidia, such conidia are effectively eliminated in immunocompetent individuals.3
 EPIDEMIOLOGY
EPIDEMIOLOGY
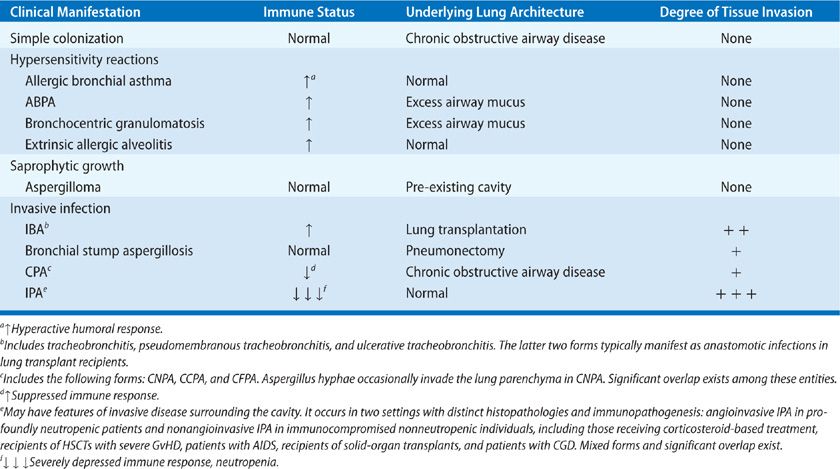
Infections by Aspergillus species cause a wide spectrum of illnesses in humans depending on the immune status of the host (Table 133-1). Hence, in immunocompetent hosts, isolation of Aspergillus spp. in respiratory secretions typically reflects colonization, not infection. Under physiologic conditions, inhalation of Aspergillus spp. does not trigger immune responses because fungal spores are covered by a hydrophobin layer that masks recognition of immunostimulatory fungal cell wall molecules by host immune cells.1–3 In atopic individuals, the fungus triggers robust immune reactions, including allergic rhinitis, asthma, hypersensitivity pneumonitis, and allergic bronchopulmonary aspergillosis (ABPA).4 In patients with pre-existing cavitary pulmonary lesions, saprophytic growth of Aspergillus spp. can lead to aspergilloma formation. Finally, in immunocompromised individuals, some Aspergillus conidia germinate into the lung to form hyphae, the invasive form of the fungus, which causes a severe, frequently fatal angioinvasive infection called invasive pulmonary aspergillosis (IPA).5–7 The degree of fungal invasion, response to antifungal therapy, and outcome of invasive aspergillosis (IA) depend on the type and severity of immunosuppression. Thus, in patients with subtle or no immune defects, chronic forms of Aspergillus infections in the lung have been described, which are characterized by an indolent clinical course with the development of progressive cavitary lesions, and minimal or no evidence of parenchymal invasion.8 A less acute form of IA, frequently called subacute IPA, has been described in patients with acquired immunodeficiency syndrome (AIDS) and those with genetic defects in NADPH oxidase (chronic granulomatous disease or CGD), whereas progressive IPA is encountered in severely immunocompromised hosts.1,2,6,7 There is no doubt that since the first description in the 1940s, IA has emerged as the major problem of modern mycology. Currently, IA is a leading cause of death in severely immunocompromised individuals, with crude mortality rates approaching 50% to 70% in patients with leukemia and recipients of hematopoietic stem cell transplants (HSCTs).5,6 In addition, invasive Aspergillus has emerged as an important pathogen in an expanding spectrum of nonneutropenic patients with chronic debilitating diseases who receive corticosteroids and/or other immunosuppressive therapies. Thus, IPA is increasingly encountered in patients with inflammatory and autoimmune diseases, chronic obstructive lung disease, and prolonged ICU stays.2 At the same time, over the past two decades, we have witnessed a significant expansion in the antifungal armamentarium with the introduction of several agents with anti-Aspergillus activity. In addition, promising new non–culture-based diagnostic methods are being developed, and considerable progress has been made in understanding the epidemiology and immunopathogenesis of IA. As a result, a significant improvement in IA-attributable mortality has been noted. As IPA currently accounts for more than 90% of invasive Aspergillus infections we devote a significant portion of this chapter to this type of pulmonary aspergillosis.
TABLE 133-1 Spectrum of Pulmonary Aspergillosis
 MYCOLOGY AND HOST DEFENSES
MYCOLOGY AND HOST DEFENSES
Of the nearly 200 Aspergillus spp., only 20 have been encountered as pathogenic in humans.1 Aspergillus fumigatus is the most frequently identified of these species; however, other species, such as Aspergillus flavus, Aspergillus niger, Aspergillus terreus, and Aspergillus nidulans have emerged as important pathogens in immunocompromised individuals. Appropriate identification of Aspergillus species is important because of differences in epidemiology, host tropism, virulence, and susceptibility to antifungal agents. For example, Aspergillus nidulans is the second most encountered mold in CGD patients, causing almost exclusively invasive infections in this specific host. In addition, certain species including A. terreus, A. flavus, and A. nidulans are clinically resistant to AMB.2,6 Recent studies using molecular identification methods discovered “cryptic” clinical isolates of Aspergillus, including Aspergillus lentulus, Aspergillus calidoustus, and Aspergillus glaucus, that had been misclassified as A. fumigatus and display inherent resistance to certain antifungal agents. Notably, A. fumigatus is nearly always the cause of allergic pulmonary disease.4 Aspergillus spp. are rapidly growing, hardy molds identified by the macroscopic appearance of the colony and by microscopic examination of the spore-bearing structures.1 Microscopically, Aspergillus spp. are characterized by the production of uniform 4- to 6-mm wide hyphae with parallel walls and distinct septa. Dichotomous branching of hyphae occurs at 45-degree angles.1 However, other filamentous fungi, such as Fusarium, Acremonium, and Scedosporium species share the same histopathological features with Aspergillus spp.1 A wide range of microscopic appearances may be observed in clinical specimens.
Sputum specimens obtained from patients and examined by direct mounting or with KOH and ink may reveal the typical 45-degree branching hyphal fragments of aspergilli, which are often associated with eosinophils and Charcot–Leyden crystals in patients with ABPA.1 Stained tissue sections reveal regular hyaline septate hyphae that are best observed with the use of periodic acid–Schiff (PAS) and Gomori methenamine silver (GMS) stain.1 Fungus ball specimens obtained from cavities connected to bronchi have hyphae that often appear to be lifeless and stain poorly. Also, although seldom seen, the fruiting heads and conidia may appear to be well formed.
 HOST IMMUNE RESPONSES
HOST IMMUNE RESPONSES
Recent years have witnessed improvements in understanding of the immunological mechanisms of host resistance to Aspergillus spp. Protection of the normal host against Aspergillus infection is mediated by a highly coordinated response that involves both innate and adaptive immunity (see also Chapters 121 and 123).3 Acquired quantitative and qualitative defects in phagocytic cell function associated with prolonged and profound neutropenia and corticosteroid-induced dysfunction of macrophages and monocytes have long been recognized as the major risk factors for IA. In addition, functional phagocytic defects due to mutations in NADPH oxidase seen in patients with CGD are associated with impaired phagocytic cell function and unique susceptibility to IA.3
Innate Immunity Consists of Three Main Effector Cell Types: (a) myeloid-derived dendritic cells that orchestrate innate and adaptive immune responses by transferring and presenting fungal antigens to T cells in resident lymph nodes; (b) pulmonary alveolar macrophages that ingest and kill inhaled conidia, primarily via oxidative reactive oxygen species (ROS)-dependent mechanisms; and (c) circulating polymorphonuclear and mononuclear cells, which are recruited to the lungs and actively participate in elimination of conidia and hyphae that escape killing by resident myeloid cells.3 Intracellular killing of Aspergillus spores within professional phagocytes occurs in acidified phagolysosomes and depends on the production of ROS. Neutrophils are unique in the ability to cause damage to Aspergillus hyphae via the release of potent antimicrobial effector molecules that are stored in specialized granules. The release of DNA filaments decorated with antimicrobial effector molecules (neutrophil extracellular traps or NETs) upon neutrophil death is a recently identified antifungal effector pathway that is efective in patients with CGD.3
In addition to professional phagocytic cells, other innate immune cell subsets appear to play important roles in Aspergillus host defense, including platelets, plasmacytoid dendritic cells, natural killer (NK) cells, and invariant NK T (iNKT) cells. These innate immune cells mediate antifungal activities either directly via the release of cytotoxic molecules or indirectly via the production of cytokines that activate professional phagocytes.3
Humoral factors also participate in the initial events of host response to Aspergillus infection. Resting conidia, germinating conidia, and hyphae are all potent activators of both the alternative and classical components of the complement cascade, promoting opsonization and chemotaxis.3 In addition, soluble C-type lectins, including mannose-binding lectins, and surfactant protein A (SP-A) and D (SP-D), which are present in alveolar fluid, enhance chemotaxis, binding, phagocytosis, and oxidative killing of conidia. C-type lectins also appear to be able to agglutinate, immobilize, and inactivate conidia.3 Importantly, alteration in SP-D and polymorphisms in mannose-binding lectin encoding genes are associated with an increased risk of chronic pulmonary aspergillosis (CPA).8 Another soluble pattern recognition receptor, the pentraxin PTX3, has been shown to bind to a range of microbial products, including Aspergillus conidia, and to confer resistance to the fungus.3 PTX3-deficient mice were susceptible to IPA because of impaired recognition of aspergilli by alveolar macrophages and dendritic cells and inappropriate induction of a Th2 response.3
In recent years, the role of adaptive immunity in Aspergillus host defense has been increasingly appreciated. Activation and maturation of dendritic cells following uptake of Aspergillus spores are critical for antigen presentation, T-cell expansion, and initiation of adaptive immunity. Activation of different pattern recognition receptors in dendritic cells by microbial ligands, including Aspergillus immunostimulatory cell wall molecules, leads to the production of distinct sets of cytokines that drive development of different Th cell subsets. Activation of toll-like receptor (TLR)-2 and TLR4 in dendritic cells has been linked to the induction of IL-12 leading to interferon-γ (IFN-γ)–producing Th1 cells, which are considered to confer broad protective immunity against fungi. In contrast, Th2 responses mediated by IL-4 lead to increased susceptibility to fungal infections. More recently, a subset of Th cells, called IL-17–producing Th (Th17) cells, has been identified and implicated in mucosal immunity against fungi. Sensing of the fungal polysaccharide β-glucan by Dectin-1 receptor on dendritic cells drives activation of antifungal Th17 responses.3 Impaired IL-17 production has been associated with increased susceptibility to IPA in mice. In humans, patients with mutations in STAT3 have selective impairment of IL-17–producing T cells and are prone to infections with Aspergillus. However, unabated Th17 responses can become detrimental for infectious outcome because of unbalanced inflammatory responses. Under physiologic conditions, Th17 cells are kept in check by IL-10–producing T-regulatory cells (T regs). However, defective ROS production in CGD is associated with compromised T reg activity, unrestrained Th17 proinflammatory responses and associated tissue pathology.3
Overall, a balanced inflammatory response is required for abrogation or effective resolution of invasive Aspergillus infection. Thus, while an early and effective proinflammatory response is required to restrict infection, an uncontrolled inflammatory response is detrimental and may eventually facilitate fungal persistence. To that end, high-level production of IL-10 by T regs may be beneficial by contributing to resolution of the inflammatory response.3 On the other side, immune tolerance may lead to adverse outcomes of infection. For example, expansion of myeloid suppressor cells that result in defective Ag presentation and Th cell priming has been described in Pneumocystis pneumonia in mice and adoptive transfer of these cells had an adverse effect on the resolution of infection. Whether a similar expansion of myeloid suppressor cells occurs in IA is currently unknown.
Immune augmentation therapies have become a top priory in fungal immunology research. Administration of Th1 cytokines has been successfully employed in the mouse model of IA. Thus, tumor necrosis factor-alpha (TNF-α), IFN-γ, interleukin (IL)-12, and IL-15 enhance effector function of professional phagocytes and have been shown to confer protection against Aspergillus infection in animal studies.3 The protective Th1 response was characterized by the presence of antigen-specific CD4+ T cells that produce IFN-γ and IL-2. Furthermore, dendritic cell–based vaccination strategies, generation of Th1/Th17 Aspergillus-specific T cells in naive mice or adoptive transfer of Aspergillus-specific CD4+ T cells in immunocompromised mice, all resulted in improved outcomes of IA in mice.2,3 Finally, restoration of immune dysfunction via cooperative activation of TLR and C-type receptor signaling resulted in cure of a chronic fungal disease of the skin in mice and could be explored as a therapeutic strategy for IA.
However, in contrast to the animal studies, in severely immunocompromised patients with IPA, immune augmentation therapies have been of unclear benefit using myeloid growth factors, Th1 recombinant cytokines or T-cell–based adoptive transfer. Thus, translating recent advances in fungal immunology to novel therapeutics against IA in humans remains a challenge. In particular, translation of results of ex vivo experiments and studies in mice with genetic defects in receptors, signaling molecules, cytokines, or chemokines into clinical practice has been difficult in view of the complex and multifactorial pathophysiology of IPA in humans. Furthermore, immune augmentation strategies and vaccination-induced protective immune therapies could be suboptimal in profoundly immunocompromised individuals. Better understanding of the immunopathogenesis of IPA at the cellular and molecular level coupled with efforts to validate research findings from mice in humans could facilitate the development of immune-based augmentation therapies.
Aspergillus virulence factors
There is debate over which factors determine Aspergillus virulence and pathogenicity.1,2 Several putative virulence factors in A. fumigatus have been identified, including various proteolytic enzymes (elastases, collagenases, trypsine), phospholipases, ribotoxin, hemolysin, gliotoxin, and many other enzymes and toxins.1,2 The mycotoxin gliotoxin in particular has received attention and been studied extensively.2 Gliotoxin exhibits immunomodulatory properties, as it has been shown to inhibit the phagocytosis of macrophages, promote apoptosis of macrophages, inhibit ROS production in human neutrophils and block B- and T-cell activation. Of interest, gliotoxin deletion mutants displayed attenuated virulence in corticosteroid-immunosuppressed mice but not in mice rendered neutropenic by treatment with cyclophosphamide, implying a direct in vivo cytotoxic effect of gliotoxin in cellular immunity.
Importantly, tissue necrosis and angioinvasion are prominent features of IA in immunocompromised patients. Of interest, gliotoxin has been shown to block angiogenesis during invasive Aspergillus growth.2,3 In fact, restoration of anti-angiogenic activity of Aspergillus via administration of pro-angiogenic factors was recently shown to potentiate activity of antifungal therapy in murine IPA. In addition, adaptation in the hypoxic environment during tissue necrosis is a prominent feature of invasive Aspergillus growth. In fact, an Aspergillus mutant deficient in a sterol regulatory element-binding protein required for hypoxia adaptation (ΔSrbA) was completely avirulent in mice.2,3 Iron metabolism is important for Aspergillus in vivo growth and mutants defective in siderophore secretion pathway fail to grow in vivo and are avirulent following infection of neutropenic mice. In addition, some have postulated that the production of catalases, superoxide dismutases, and mannitol by Aspergillus may protect the fungus from oxidative damage induced by phagocytic cells.2,3 Moreover, the melanin pigment and rigid protein coat layer composed of hydrophobic rodlet fascicles on the Aspergillus conidial surface may confer resistance to phagocytosis.2 In fact, melanin may act as a virulence factor by masking surface immunostimulatory molecules, blocking ROS production and acidification of fungal phagosomes. On the other hand, recent evidence from comparative genomic analysis across filamentous fungi suggests that A. fumigatus virulence results from the immunosuppression or possibly genetic susceptibility of the host rather than from specific, unique fungal determinants.2,3 Hence, A. fumigatus appears to be a saprotrophic fungus that only becomes pathogenic for very simple biological reasons: it is present in high concentrations in the atmosphere, it grows faster than any other airborne fungi at physiologic mammalian temperatures (above 37°C), and it can overcome the defense of the host not because of specific virulence mechanisms but because of inefficient host immune responses. Taken together, these data suggest that approaches targeted at augmenting cellular immunity, such as neutralization of Th2 cytokines, enhancement of Th1 responses, and dendritic-cell vaccine-based vaccination warrant investigation as therapeutic modalities for invasive Aspergillus infections.2,3
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
The varied clinical manifestations of disease related to Aspergillus species are considered in detail below.
Hypersensitive Reactions
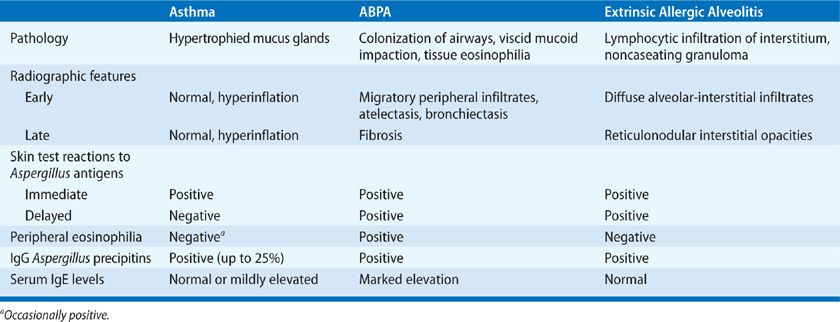
Hypersensitivity lung diseases that result from exposure to A. fumigatus allergens include allergic asthma, ABPA, and hypersensitivity pneumonitis.4,6 In atopic individuals, inhalation of Aspergillus spores triggers an IgE-mediated allergic inflammatory response in the bronchial mucosa, leading to excessive mucus production, bronchial obstruction, and asthma.4,6 Similarly, ABPA develops following sensitization to A. fumigatus allergens in a unique subset of atopic individuals and patients with cystic fibrosis (CF), who are genetically susceptible for ABPA.4 The immune response to Aspergillus antigens in both patients with allergic asthma and those with ABPA is characterized by a robust Th2 polarized CD4+ response.
In nonatopic individuals, persistent airway colonization and, more rarely, a type of hypersensitivity pneumonitis called extrinsic allergic alveolitis can occur as a result of massive or repeated inhalation of Aspergillus conidia.4,8 Hypersensitivity pneumonitis in these patients manifests with dyspnea because of pulmonary constriction and an influenza-like syndrome with fever, malaise, and fatigue. In contrast to other allergic diseases caused by A. fumigatus, hypersensitive pneumonitis is characterized by extensive infiltration of neutrophils in the acute phase as a result of a Th1 CD4+ response.4,8 Early recognition of these syndromes is important for early initiation of appropriate treatment to prevent progression to permanent lung damage. Table 133-2 summarizes the hypersensitivity syndromes that result from exposure to Aspergillus.
TABLE 133-2 Hypersensitive Reactions to Aspergillus
Simple Colonization
Although there is not a uniform definition of colonization, it should be considered in cases of isolation of Aspergillus spp. from cultures of the respiratory tract in patients without evidence of invasive or allergic disease.8 Patients with structural lung diseases such as chronic obstructive pulmonary disease (COPD) are at increased risk for persistent Aspergillus colonization. Tissue invasion by aspergilli is not a feature of saprophytic colonization, although IPA following subsequent dysfunction of host defenses may develop. In fact, Aspergillus colonization has been shown to be a marker for the development of IA in immunocompromised individuals, particularly lung and bone marrow transplant recipients, and may precede invasion for up to 3 months.8 Quantitative real-time polymerase chain reaction (PCR) and measurement of BAL Aspergillus galactomannan antigen are both highly sensitive and specific assays in distinguishing Aspergillus colonization from invasive infection.2,6
Allergic Bronchial Asthma
Asthmatic patients may become sensitized to Aspergillus conidia as a consequence of thick bronchial secretions that trap fungal spores.4,8 These spores seldom germinate in the bronchial airways. Allergic bronchial asthma develops in patients who are atopic and is perpetuated by inhalation of Aspergillus antigens, which typically causes acute bronchospasm. The presence of transient infiltrates has been described during the immediate reaction but is not frequent. As is typical in asthma of other etiologies, eosinophils and serum IgE antibody are increased. Immediate skin reactions to Aspergillus antigens are positive, but specific precipitating antibodies (IgG) are usually negative.4,8 Avoidance of exposure to Aspergillus spores can diminish the frequency and severity of bronchospasm in this group of patients. Aspergillus bronchitis is a rare, distinct clinical entity that has been recently described in immunocompetent individuals with structural lung disease, chronic pulmonary symptoms, and lack of response to antibiotic therapy. All of these patients had evidence of Aspergillus growth in their lungs, marked improvement following antifungal therapy and did not fulfill criteria for asthma or other forms of invasive or chronic aspergillosis.
Allergic Bronchopulmonary Aspergillosis
The important clinical entity of allergic bronchopulmonary aspergillosis (ABPA) is discussed below. The reader is also referred to Chapters 48 and 71.
Immunopathogenesis and Risk Factors It has been recently proposed that ABPA develops in genetically susceptible patients with asthma or CF because of increased activity of A. fumigatus-specific Th2 CD4+ cells (see also Chapter 48).4 Hence, in addition to a history of atopy, predisposing factors for ABPA include immunogenetic human leukocyte antigen (HLA)-distinct phenotypes (e.g., HLA-DR 2- and HLA-DR 5-specific alleles), mutations in the CF transmembrane conductance regulator (CFTR) gene, polymorphisms of the collagen region of the SP-A2, and/or other collections (e.g., mannose-binding lectins).4,8 In addition, particular physicochemical characteristics of respiratory secretions and a history of environmental exposure may play a role in ABPA pathogenesis.4,8 The role of newly identified Th17 pathway, which has a critical role in inflammation via neutrophil recruitment has not been extensively evaluated in ABPA. However, in a mouse model of asthma, Dectin-1 dependent IL-22 production was important for disease development. In addition, TNF-α–producing inflammatory DCs have been recently shown to be important regulators of IL-17–producing T cells and neutrophilia in a mouse model of ABPA.
In patients with ABPA, inhaled A. fumigatus conidia are able to colonize, persist, and germinate, leading to growth of hyphae in mucus plugs, which are often observed in expectorated sputum.4 During this process, A. fumigatus releases a panel of proteases and other toxins that are capable of further compromising mucociliary clearance, breaching the airway epithelial barrier, and activating the immune system in the lung.4
IgE molecules on the surface of mast cells initially recognize A. fumigatus antigens. Binding of these antigens with IgE causes mast-cell degranulation, releasing mediators that cause vasodilatation and vascular leakage so that serum components, including anti-Aspergillus IgG, enter the bronchi and combine with Aspergillus antigens to form IgG-containing immune complexes.4 These complexes activate the complement cascade leading to inflammation and pulmonary damage. Importantly, investigators have identified specific Aspergillus antigens capable of inducing IgE and IgG responses in patients with ABPA but not in patients who did not have ABPA, implying that an aberrant host response occurs in patients with ABPA.4
The key element in the immunopathogenesis of ABPA appears to be an abnormal T-lymphocyte cellular immune response to A. fumigatus conidia. Hence, A. fumigatus allergens released by mycelia are processed by antigen-presenting cells bearing HLA-DR 2 or HLA-DR 5 and presented to T cells within the lymphoid tissue of the lung.4,8 The T-cell response to Aspergillus allergens is then shifted toward a Th2 CD4+ cell response with synthesis and secretion of the cytokines IL-4, IL-5, and IL-13.4,8 In addition, production of chemotactic cytokines (chemokines) by inflammatory cells, including monocyte chemoattractant protein-1 (MCP-1/CCL2), eotaxin (CCL11), IL-8, and macrophage inflammatory protein-1α (MIP-1α/CCL3) appears to promote the Th2 cell response.4,8 In addition, T cells, B cells, NK cells, and eosinophils may be hyperresponsive to IL-4.4,8 This leads to a positive-feedback amplification loop of Th2 CD4+ cells, synthesis of IL-4, and polyclonal activation of CD23+ CD86+ B cells.4,8
Clinical Features ABPA is a hypersensitivity disease of the lung that is virtually almost always related to A. fumigatus (discussed in detail in Chapter 48). Some key points are highlighted here. Seven percent to 14% of patients with poorly controlled asthma and 7% to 9% of those with CF meet the diagnostic criteria for ABPA.4 In fact, nearly all patients with ABPA have a history of chronic asthma. Rarely, patients present without symptoms or physical findings despite having radiographically detected abnormalities. Clinically, ABPA manifests with episodic wheezing, malaise, low-grade chronic fevers and cough, sputum containing brown flecks and plugs, chest pain, pulmonary infiltrates, and sputum and blood eosinophilia.4,8 Patients may have a history of recurrent pneumonias and frequent use of antibiotic therapy. Mucus plugs form in the proximal bronchi and can progress to mucoid impaction, resulting in atelectasis with transient pulmonary infiltrates. Mucus plugs often yield aspergilli in culture (Figs. 133-1 and 133-2). As ABPA progresses, central bronchiectasis becomes a dominant feature of the disease and may result in chronic bronchorrhea and occasionally hemoptysis, development of clubbing, and fixed, characteristic radiographic abnormalities.4,8 In patients with CF in particular, ABPA often manifests with hemoptysis and may be complicated by pneumothorax.4
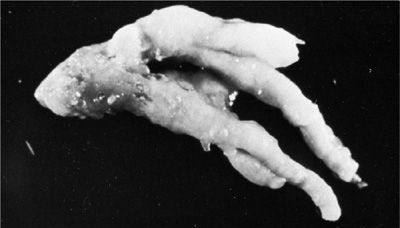
Figure 133-1 Mucus plug expectorated by a patient with ABPA. Of note is the tapering, cylindrical shape with branching characteristic of the parent bronchi. (Used with permission of Dr. Frederic Askin, Chapel Hill, NC.)
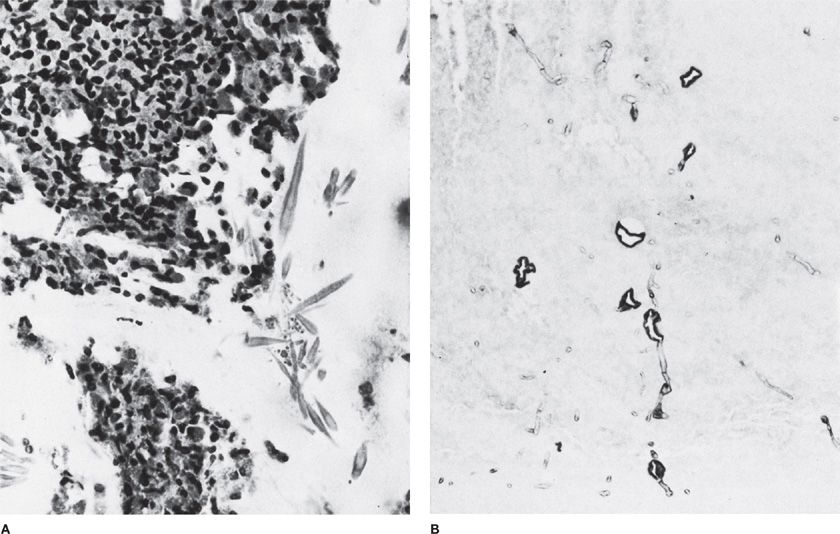
Figure 133-2 High-magnification photomicrograph of inspissated mucus in mucoid impaction of the bronchus. A. Groups of Charcot–Leyden crystals adjacent to clusters of necrotic eosinophils. H&E stain, ×520. B. Aspergillus hyphae. Of note is the transition from regular, thin septate hyphae to dilated, often folded degenerating forms. H&E stain, ×354. (Used with permission of Drs. S. Albelda and G.H. Talbot.)
Radiographic Findings The radiographic features of ABPA have an essential role in diagnosis, as they may distinguish ABPA from simple, chronic asthma. Pulmonary infiltrates in cases of ABPA are initially transient, but they ultimately progress to permanent radiographic changes.4,8 In many instances, the “ring sign” indicative of bronchial wall thickening, “parallel” shadows or “tram lines” suggestive of bronchiectasis, and branching “finger-in-glove” opacification of the dilated bronchi because of mucoid impaction are seen on regular chest radiographs of patients with ABPA (Fig. 133-3).4,8 The presence of proximal bronchiectasis (Figs. 133-3 and 133-4), which is characterized by normal filling of bronchi distal to the sacular bronchial lesion, is considered a hallmark for the diagnosis of ABPA.4 ABPA lesions are either focal or bilateral and tend to occur more frequently in the upper lobes. Late radiographic findings in patients with ABPA include cavitation, local emphysema, contracted upper lobes, and honeycomb fibrosis.
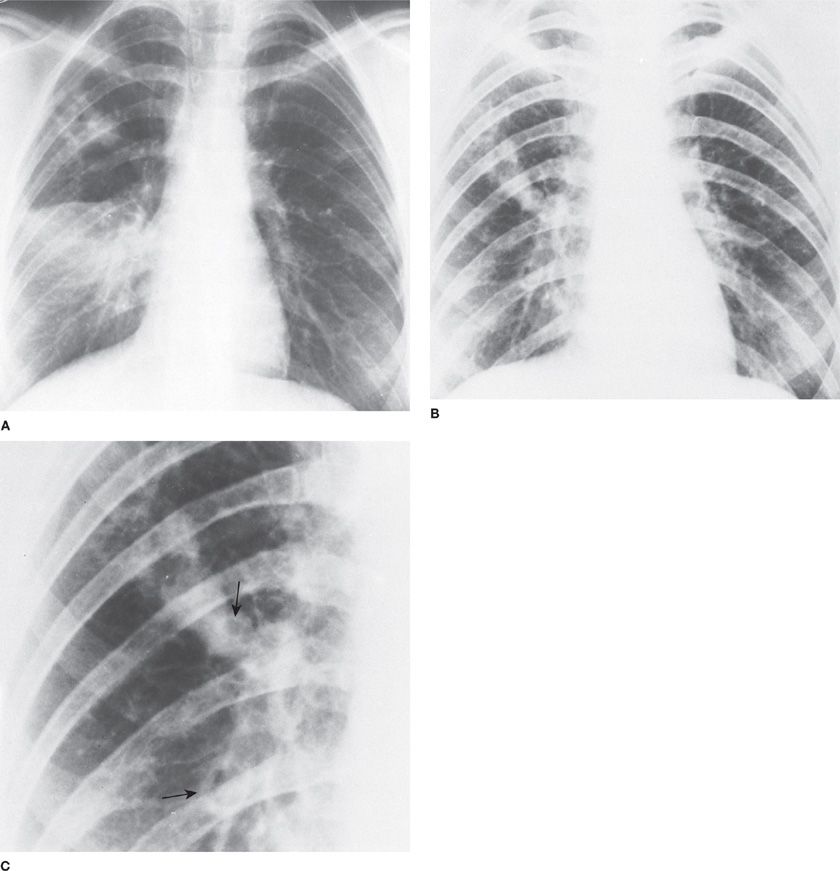
Figure 133-3 A 23-year-old woman with documented ABPA. A. Chest radiograph showing mucoid impaction in the right upper lobe and alveolar consolidation in the right middle lobe. B. Eight months later, the alveolar consolidation has resolved, and the appearance of the mucoid impaction in the right upper lobe has changed. C. Close-up of the right lung in B showing the “gloved finger” appearance of mucoid impaction as well as ring shadows (arrows) characteristic of bronchiectasis. (Used with permission of Drs. S. Albelda and G.H. Talbot.)
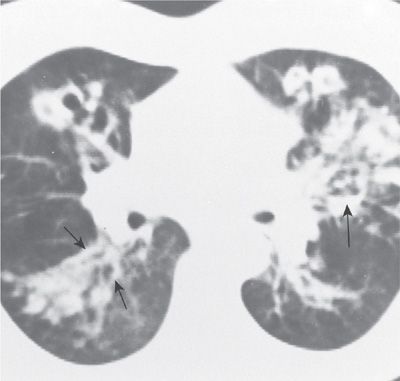
Figure 133-4 Proximal saccular bronchiectasis characteristic of ABPA. The CT scan reveals multiple rounded, dilated bronchi (small arrows). Of note is the air–fluid level (large arrow). (Used with permission of Drs. S. Albelda and G.H. Talbot.)
Chest computed tomography (CT) has been a significant aid in diagnosing ABPA because it is more sensitive than regular chest radiography; however, there are no pathognomonic CT findings for this entity.4,8 Besides central bronchiectasis, CT findings highly suggestive of ABPA include the presence of varicose or cystic bronchiectasis of segmental and subsegmental bronchi (90% in ABPA vs. 30% in simple asthma), mucoid impaction of the segmental and subsegmental airway (67% in ABPA vs. 4% in simple asthma), and small airway abnormalities such as centrilobular nodules and tree-in-bud opacities (93% in ABPA vs. 28% in simple asthma).4,8 In patients with CF especially, CT diagnosis of ABPA is particularly challenging, as there is a significant overlap in the radiographic features of both entities.4 In fact, the presence of high-attenuation mucus plugs because of dystrophic calcification is the only CT finding that favors the diagnosis of ABPA in patients with CF.
Diagnosis The “classic” patient with ABPA fulfills the criteria listed in Table 133-3. It has been suggested that the minimal essential criteria for the diagnosis of ABPA include the presence of (a) asthma, (b) immediate cutaneous reactivity to Aspergillus antigens, (c) elevated total serum IgE levels (>1000 U/mL), (d) elevated serum IgE and IgG levels to A. fumigatus, and (e) central bronchiectasis.4,8 However, asymptomatic pulmonary involvement occurs in patients with ABPA and may lead to diagnostic uncertainty, delayed diagnosis, and irrevocable structural lung damage. Furthermore, corticosteroid-based therapy for asthma may mask the signs of ABPA, leading to a progressive decline in pulmonary function. Because the above criteria may not apply in such cases, some have proposed that serological evidence (all three serological tests listed in Table 133-3 and immediate skin reactivity test to Aspergillus antigens need to be positive) even without the presence of bronchiectasis may be sufficient to support an early diagnosis of ABPA.4,8 In addition, the diagnostic criteria for ABPA in patients with CF were recently modified, because of the overlapping features between these entities. The new criteria consist of (a) clinical and/or pulmonary function deterioration from baseline status, (b) positive immediate cutaneous reaction to A. fumigatus antigens or elevated IgE A. fumigatus antibody serum level, (c) elevated serum total IgE level >1000 U/mL, (d) elevated serum IgG A. fumigatus antibody level or positive A. fumigatus precipitins, and (e) abnormal chest imaging findings or a change in baseline abnormalities.4
aTest must be positive for serological diagnosis in asymptomatic patients.
Source: Adapted with permission from Rosenberg M, Patterson R, Mintzer R, et al., Clinical and Immunological criteria for the diagnosis of allegic bronchopulmonary aspergillosis. Ann Intern Med. 1977;86:405–414.
Important clinical clues that should alert clinicians to the possibility of ABPA include refractory asthma or asthma with any of the following features: bronchiectasis, radiographic infiltrates, prominent eosinophilia, and expectorated brown plugs that may contain eosinophils or hyphae (Fig. 133-2).4 In addition, in patients with CF, prominent wheezing is considered to be suggestive of ABPA. Rarely, ABPA may be caused by non-fumigatus aspergilli or other fungi; in such cases, the serological tests will be negative.
Because no single clinical or immunological feature is diagnostic for ABPA, this disease is underdiagnosed. Until recently, serological diagnosis of ABPA was based on Aspergillus crude extracts, which are cumbersome, lack reproducibility, and frequently cross-react with other antigens. However, recent studies indicate that standardized recombinant A. fumigatus allergens have the potential to substantially increase the specificity and sensitivity of diagnosis of A. fumigatus-related diseases, including ABPA.4,8 Several recombinant A. fumigatus allergens, including Aspf1, Aspf2, Aspf3, Aspf4, Aspf6, and Aspf16, show high specificity for the detection of sensitization to A. fumigatus and for the diagnosis of ABPA and appear to be superior to crude Aspergillus allergen extracts.4 However, because the sensitivity of each recombinant A. fumigatus allergen is suboptimal, use of a panel of recombinant allergens is favored to increase the sensitivity of the diagnosis of ABPA. This diagnostic approach remains investigational. Further standardization and development of commercially available immunoassays containing a combination of these allergens holds promise for improving the diagnosis of ABPA.
Because patients with CF are at high risk for ABPA, initiation of annual screening in patients with >6 years of age for ABPA with monitoring of serum total IgE levels has been recommended with further diagnostic workup for ABPA in cases with IgE levels >500 U/mL.4 Likewise, because treatment with corticosteroids has a significant impact on disease progression, screening CT has been advocated for asthmatics with a skin-prick hypersensitivity test positive for A. fumigatus extracts to identify ABPA at an early stage.4,8
Histopathology Because the diagnosis of ABPA is based on clinical and laboratory criteria, use of lung biopsy to confirm the diagnosis is usually not required. The presentations of the hypersensitivity syndrome associated with fungi are discussed in detail in Chapter 48. Some of the histopathological presentations associated with ABPA overlap those of invasive Aspergillus infection. Thus, eosinophilic infiltrates may be present in patients with a chronic invasive Aspergillus infection as well as in those with other allergic lung entities, such as Löffler syndrome, tropical eosinophilic pneumonia, and chronic eosinophilic pneumonia (see Chapters 71 and 137).4,8 Mucoid impaction associated with ABPA or other fungi may predispose patients to bacterial superinfection (Figs. 133-1 and 133-2). Necrotizing granulomas in association with the presence of Aspergillus spp. in asthmatic patients may be observed and is considered part of the ABPA syndrome.4,8 The histopathological distinction between ABPA, with fungal elements restricted to the airways, and semi-invasive or fully IA may be obscured by tissue necrosis or poor granuloma formation. These presentations are discussed further below and in Chapter 51.
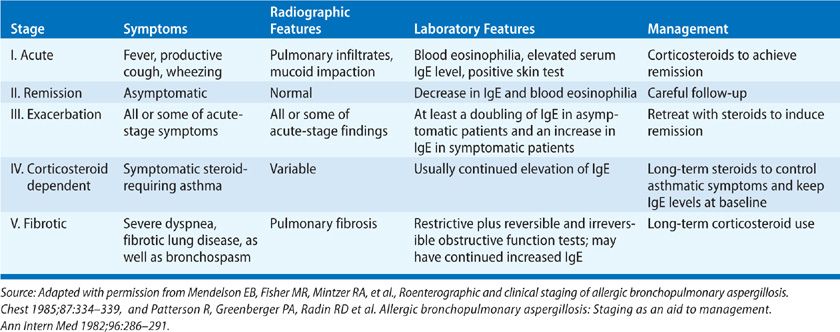
Treatment ABPA may progress through five stages based on clinical, serological, and radiographic findings, which are summarized in Table 133-4.4,8 Importantly, the first four stages are potentially reversible without long-term sequelae; only the last stage, which is characterized by fibrosis and bronchiectasis, is irreversible. The goals of therapy for ABPA are to treat acute exacerbations of disease and prevent the development of fibrosis and bronchiectasis. Therefore, early diagnosis of ABPA before permanent lung damage occurs is critical. Therapeutic modalities for ABPA include oral corticosteroids, metered-dose inhaler medications, and systemic antifungal agents.4,8
TABLE 133-4 Staging System for Allergic Bronchopulmonary Aspergillosis (ABPA)
Use of oral corticosteroids remains the cornerstone of the treatment of ABPA.4,8 These agents suppress the inflammatory response provoked by Aspergillus infection rather than eradicate the fungus. Recommended corticosteroid doses for acute exacerbations of ABPA are 0.5 to 1.0 mg/kg of prednisone equivalent daily for 1 to 2 weeks, followed by 0.5 mg/kg per every other day for 6 to 12 weeks. The physician should then attempt to taper the patient’s corticosteroid dosage. However, some patients cannot be successfully weaned off corticosteroids and may require a daily maintenance dose of at least 7.5 mg.
Increasingly, use of itraconazole, an azole antifungal with activity against Aspergillus spp., has been employed as adjunctive fungal antigen-reducing therapy for ABPA, especially in patients with frequent relapses or corticosteroid dependence.4,8 A randomized controlled study reported that oral administration of itraconazole (200 mg twice daily) was beneficial in terms of significant improvement of symptoms, pulmonary function, chest radiographic findings, and serum IgE levels in asthmatic patients with ABPA; uncontrolled studies suggested a similar benefit in patients with CF and ABPA.4,8 However, use of itraconazole requires determination of drug levels to ensure adequate absorption and poses significant issues with long-term tolerability and drug–drug interactions.5,6 Another important issue of prolonged itraconazole use is the emergence of azole-resistant Aspergillus isolates. In addition, itraconazole has a 40% failure rate in ABPA treatment.6 The newer triazole voriconazole has improved fungicidal activity against Aspergillus spp. and better bioavailability when compared with itraconazole.5,6 A recent retrospective study in 20 ABPA patients who failed or were intolerant to itraconazole and received either voriconazole or posaconazole found sustained clinical and immunologic responses and reduced corticosteroid requirements at 12 months in over 75% of treated patients.5–7 However, high relapse rates (up to 57%) have been reported following discontinuation of azole treatment in patients with ABPA, and there are concerns for the development of secondary resistance in Aspergillus following prolonged courses of antifungal therapy.
Use of inhaled corticosteroids may help control the symptoms of asthma but does not prevent episodes of eosinophilic infiltration and mucus impaction and is generally thought to have no influence on progressive lung damage.4,8 Inhaled bronchodilators such as β agonists, anticholinergics, and leukotriene antagonists may be used to treat asthma-related symptoms.4,8 Inhaled antifungal agents such as nystatin and amphotericin B deoxycholate (AMB-D) may offer temporary suppression of colonization, but penetration of these agents into plugged bronchi is limited and recolonization rapidly occurs once therapy ends. Meticulous bronchial toilet is important for clearance of A. fumigatus from the airway. Thus, physical therapy with postural drainage is an important adjunctive treatment. Increased hydration, use of expectorants, and in selected patients, bronchial lavage may aid in viscid mucus clearance. Avoidance of environmental reservoirs of Aspergillus spp. such as compost heaps, grain silos, and decayed organic matter may help prevent exacerbation of ABPA. In patients with CF ABPA, differentiating between a bacterial exacerbation of CF and an ABPA flare is important, because initiation of corticosteroids may have detrimental effects in patients with bacterial infection; a serological assessment is essential as part of this evaluation.4
Immunotherapy with the injection of conventional allergens has not been shown to be of value for ABPA.4,8 However, newer immunomodulatory approaches, including immune modulation toward a Th1 response by using A. fumigatus synthetic peptides, DNA-based vaccines, and immunostimulatory molecules such as cytosine polyguanine oligonucleotides, are being explored in animal models with promising results.
Monitoring of treatment efficacy and toxicity is imperative. Long-term follow-up of patients with ABPA has shown that increasing serum levels of IgE is often a harbinger of a clinical flare of the disease and that the levels of IgE decline correlate with a clinical response to corticosteroids.4,8 Thus, a prudent course of action is to determine baseline serum IgE levels when ABPA has been controlled by corticosteroid-based therapy and then monitor serum IgE levels regularly (every 1—2 months). Total serum IgE levels usually should decrease by at least one-third within 6 weeks from the initiation of corticosteroid-based treatment; infiltrates should be resolved after 1 to 2 months, and the results of pulmonary function tests should improve. Although serum IgE levels seldom return to normal, reinstitution of corticosteroids should be considered even in asymptomatic patients if the serum IgE level is double that of the baseline value.4,8 In patients with symptomatic ABPA, chest radiographs should be obtained every 3 months during the first year of follow-up and yearly thereafter; pulmonary function tests should also be performed yearly.4,8 However, no one has identified certain prognostic indicators for progression or regression of ABPA, and whether “pre-emptive” treatment of asymptomatic flares alters the natural history of the disease remains controversial.
Bronchocentric Granulomatosis
Bronchocentric granulomatosis is a rare hypersensitivity syndrome that is characterized histologically by replacement of bronchial mucosa with necrotizing granulomatous tissue.8 Also, eosinophilic infiltration of bronchioles and fibrosis is prominent, whereas there is no evidence of Aspergillus invasion. Aspergillus hyphae have been demonstrated within the lesions in approximately half of the cases of bronchocentric granulomatosis. This syndrome is associated with asthma in half of the patients described and likely represents a severe exuberant focal manifestation of ABPA rather than the more generalized pulmonary pathology evident in patients with ABPA.8 Patients typically present with chronic symptoms, such as malaise, cough, low-grade fever, dyspnea, dull chest pain, and hemoptysis associated with a focal lesion on chest radiographs, often in an upper lobe. Occasionally, radiographic findings of multiple nodular or mass-like lesions simulate metastatic carcinoma.8 Diagnosis of bronchocentric granulomatosis is made based on biopsy or often retrospectively after removal of the lesion, which is curative. Some patients, especially those with multifocal lesions, may need corticosteroid-based therapy for ABPA.8
Extrinsic Allergic Alveolitis
Heavy or repeated exposure to Aspergillus conidia and mycelia may result in a hypersensitivity reaction affecting the alveoli in nonatopic individuals known as extrinsic allergic alveolitis (see Chapter 58).8 Malt workers, distillers, brewers, and others exposed to moldy straw or grain have suffered attacks consisting of cough, dyspnea, fever, chills, myalgia, and malaise 4 to 8 hours after exposure to Aspergillus antigens.8 Repeated exposures may lead to malt worker’s lung or farmer’s lung or to the development of granulomatous disease or interstitial fibrosis. The immunopathogenesis of extrinsic allergic alveolitis involves cell-mediated immunity (type IV response) and immune complex deposition (type III response) and likely involves an intricate interaction between these immune mechanisms.8
Radiographic changes in the acute syndrome include diffuse alveolar-interstitial infiltrates that may resolve with the removal of the inciting antigen. Patients with chronic extrinsic allergic alveolitis may have fine reticulonodular interstitial infiltrates that may progress to pulmonary fibrosis with honeycombing.8 Serum IgG antibodies (precipitins) against Aspergillus antigens are present in patients with extrinsic allergic alveolitis; however, the serum IgE level is typically normal. Skin tests usually demonstrate an Arthus reaction at 4 to 8 hours and occasionally may be preceded by an immediate wheal and flare reaction and followed by a delayed reaction (36–48 hours afterward). Removal or avoidance of the source of antigen exposure is crucial in the management of extrinsic allergic alveolitis, as spontaneous recovery often ensues once exposure has ended.8 Administration of corticosteroids is helpful in aiding the resolution of acute symptoms and reduces the likelihood of structural damage. However, corticosteroids are not helpful once fibrosis has developed.
Aspergilloma
Saprophytic colonization of a parenchymal lung cavity by Aspergillus is referred to as aspergilloma, mycetoma, or a fungus ball.8 A fungus ball consists of both dead and living mycelial elements, fibrin, mucus, amorphous debris, inflammatory cells, and degenerating blood and epithelial elements. The mycelial mass may lie free within the cavity or be attached to the cavity wall by granulation tissue. Spontaneous shrinkage or disappearance of aspergilloma has been reported in 7% to 10% of cases and is often associated with bacterial superinfection of the cavity.8 Aspergillomas only rarely increase in size. Most aspergillomas are caused by A. fumigates, but some, especially in patients with diabetes mellitus, are caused by A. niger, in which case oxalic acid crystals may be seen in the sputum.8 Rare cases of extrapulmonary sites of aspergillomas involving the skin, brain, retroperitoneal area, and sinuses have been described.
Pathophysiology The pathogenesis of aspergilloma usually involves colonization and proliferation of the fungus in a pre-existing pulmonary cavity (secondary aspergilloma). The cause of a pre-existing cavity is most commonly prior cavitary tuberculosis; aspergilloma has been reported in 11% to 17% of patients with post-tuberculosis cavities.8 In fact, 20% of patients who recover from cavitary tuberculosis develop aspergillomas within the next 3 years. Of interest, in developing countries tuberculosis is the predisposing factor for over 90% of cases of aspergilloma. However, an aspergilloma may complicate a wide spectrum of cavitating pulmonary diseases, such as sarcoidosis, histoplasmosis, blastomycosis, AIDS (especially in cases of atypical Pneumocystis jiroveci pneumonia), pneumonia and/or lung abscess, pulmonary or bronchial cysts, bronchiectasis, pulmonary fibrosis, asbestosis, adenocarcinoma, ankylosing spondylitis, rheumatoid nodules, cyanotic heart disease, and pulmonary infarction.8
Primary aspergilloma, which arises within the bronchial tree with the proliferation of Aspergillus leading to a pulmonary cavity, is far less common. The clinical conditions leading to the initiation of a cavitary process and formation of a fungus ball include IPA, chronic necrotizing pulmonary aspergillosis (CNPA), and ABPA. IPA may lead to primary aspergilloma during bone marrow recovery, as the host is able to mount an inflammatory response and ward off the fungus. Pneumothorax may be a severe complication of pulmonary mycetoma that is reported as a rare occurrence in patients with hematological malignancies.8 ABPA may cause bronchiectasis in the chronic phase of the disease and result in aspergilloma secondary to growth of a fungus distal to a plugged bronchus.4,8 In fact, aspergillomas have been found in 7% of patients with ABPA in one study. Thus, aspergilloma may provide a stimulus for the perpetuation of ABPA.
Typical biofilm structures have been observed in aspergillomas from patients and in a murine model of IPA. Aspergilloma formation is linked with the ability of the fungus to form an extracellular hydrophobic matrix with typical biofilm characteristics under different static conditions including interaction with bronchial epithelial cells. This extracellular matrix is composed of galactomannan, α-1,3-glucans, monosaccharides and polyols, melanin and proteins including major antigens and hydrophobins. In particular, the presence of α-1,3-glucans in the extracellular matrix has a predominant role in the agglutination of Aspergillus germinating spores and hyphae and the subsequent aspergilloma formation. Melanin production during infection could also play a role in facilitating lung tissue invasion possibly by attenuating or evading immune response. Defective extracellular matrix penetration by most antifungal agents makes them significantly less effective when A. fumigatus is grown under biofilm versus planktonic conditions. Thus, chronic exposure and suboptimal azole concentrations in fungal biofilms could explain the emergence of azole resistant A. fumigatus isolates reported in these patients.2,3,8
Clinical Features/Diagnosis Hemoptysis is the typical manifestation of aspergilloma, as it is seen in 50% to 90% of patients.8 Hemoptysis is typically infrequent and mild but can be occasionally massive or even fatal. The presence of any other symptoms, including dyspnea, fever, malaise, and weight loss, in patients with aspergilloma should be attributed to the underlying cavitary pulmonary disease or bacterial superinfection or suggest the presence of an overlapping IPA syndrome such as CNPA. The diagnosis is usually made based on clinical and chest radiographic features coupled with serological evidence of precipitating antibodies to Aspergillus spp.
Chest radiographs reveal a solid round mass within a cavity (3–5 cm diameter) partially surrounded by a radiolucent crescent (Monod’s sign) (Fig. 133-5).8 Movement of the fungus ball within the cavity may be appreciated when comparing upright and decubitus images. A solitary lesion in the upper lung fields is the most common radiographic feature of aspergilloma, as pre-existing tuberculosis cavities is the most common predisposing condition. However, aspergillomas can be bilateral and multiple. In many cases, chest CT may be helpful in further delineating the radiographic features of an aspergilloma that are not apparent on chest radiographs. On CT scans, globules of gas are often seen within the hyphal mass. CT angiography may also provide useful information for patients with hemoptysis by identifying hypertrophic bronchial arteries that often supply the cystic wall of aspergillomas.4,8
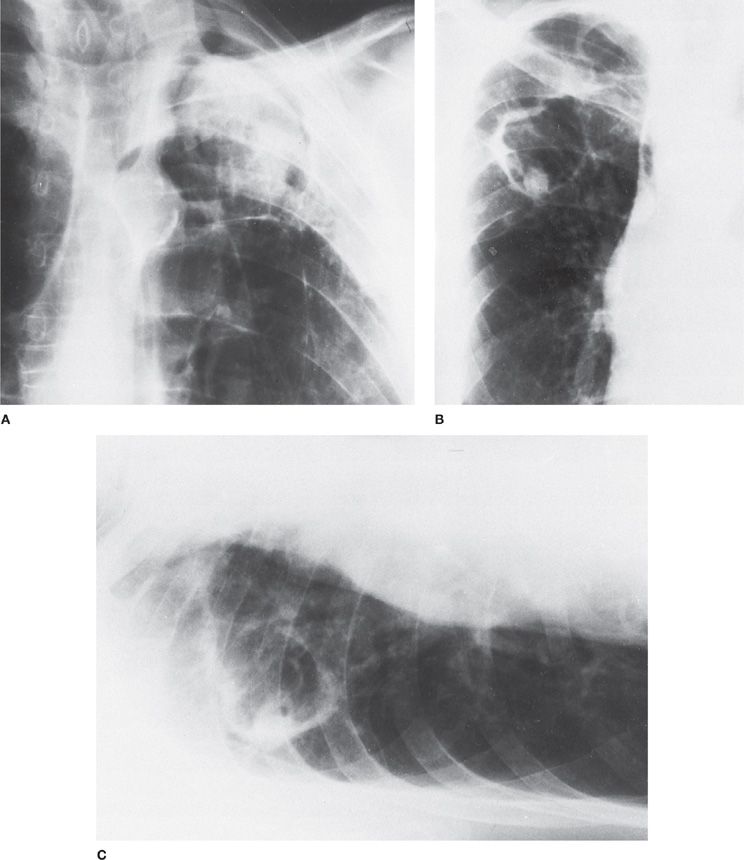
Figure 133-5 Radiographic appearances of pulmonary aspergillomas. A. A 51-year-old man with a history of tuberculosis showing the classic crescent-shaped patch of air (Monod’s sign) as well as marked pleural thickening. B. A 49-year-old man with ankylosing spondylitis in whom an aspergilloma developed inside a thin-walled cavity. C. Decubitus film of the patient in B showing a fungus ball that moved when the patient changed position. (Used with permission of Drs. S. Albelda and G.H. Talbot.)
Although sputum cultures are positive for Aspergillus in more than half of all patients with aspergilloma, culture is not a sensitive and specific diagnostic marker.8 Precipitating antibodies to Aspergillus antigens are present in the sera of more than 95% of patients with aspergilloma; however, some patients receiving corticosteroids may be seronegative.8 Eosinophilia, IgE, and skin-test reactivity may be seen in individuals who are allergic to the fungus, but these are not consistent findings. The main challenge for the clinician is to differentiate aspergilloma from other lung conditions, including lung cancer, cavitating Wegener granulomatosis, a blood clot in a pre-existing cavity, a disintegrating hydatid cyst, and a pulmonary abscess, all of which often share radiographic features with aspergilloma. Therefore, a positive precipitin reaction for Aspergillus is particularly helpful in establishing the diagnosis of aspergilloma. Clinical judgment and a low threshold for performing a lung biopsy in atypical cases are required.
Predictors of poor prognosis for aspergilloma include a progressive increase in the size and/or number of aspergillomas, severe underlying lung disease with a poor lung reserve, immunosuppressive therapy, AIDS, sarcoidosis, a rising Aspergillus-specific IgG titer, and repetitive episodes of severe hemoptysis.4,8
Treatment There is no consensus on the treatment of aspergilloma because of a lack of controlled studies. Because life-threatening hemoptysis occurs only in a minority of patients, subjecting all patients with aspergilloma to surgical therapy, which is often associated with significant morbidity and mortality, seems to be inappropriate.8 Management options for aspergilloma currently include systemic or local administration of antifungal agents, surgical resection, and conservative management with careful follow-up without specific medical or surgical intervention.8 Often, the best course of action for asymptomatic patients with aspergilloma is carefully repeated clinical evaluation with periodic chest radiographs without surgical intervention. Therapeutic considerations must include the individual patient’s health status with attention to the potential risks of each treatment.
The definitive treatment of aspergilloma is surgical resection. However, in many patients, surgery is contraindicated because of severe underlying pulmonary dysfunction; the operation per se is associated with significant mortality and serious postoperative complications such as hemorrhage, bronchopleural fistula, bacterial superinfection, and empyema.6,8 Therefore, it has been suggested that surgical resection of aspergilloma should be restricted to patients with severe, life-threatening hemoptysis and preserved pulmonary function. Surgical resection should also be considered for patients with poor prognostic features (e.g., chronic immunosuppression, sarcoidosis, and increasing Aspergillus-specific IgG titers).6,8 Extrapleural resection has been reported to improve outcome. For patients who are unfit for surgical resection, an alternative approach is cavernostomy, which is performed under local anesthesia; however, cavernostomy is also associated with mortality and mediocre results.6,8 This procedure should be considered as a last resort.
Intracavitary instillation of an antifungal agent is a promising alternative treatment in patients with severe pulmonary dysfunction who are poor candidates for surgery. CT-guided percutaneous instillation of AMB-D has been shown to be effective for aspergilloma in several cases of massive hemoptysis, with resolution of hemoptysis within 5 days.8 The response to percutaneous injection of AMB-D was sustained with no recurrences for several months, improvement or even resolution of radiographic abnormalities, and reduction of serum Aspergillus antibody titers.4,6,8 Endobronchial instillation of ketoconazole via fiberoptic bronchoscopy has also been successful.8 Overall, topical therapy with antifungal agents is ideal for patients with a solitary aspergilloma who have severe hemoptysis and contraindications to surgical resection.
Bronchial arterial embolization (BAE) has been extensively used in the management of hemoptysis in patients with aspergilloma.4,8 However, this approach has proven to be only temporarily effective, and recurrence of hemoptysis usually occurs because of the presence of collateral vessels in the involved area. Hence, BAE seems to be appropriate only as a “bridge” procedure in patients with massive hemoptysis until surgical resection of the aspergilloma can be performed. Also, radiation therapy has been shown to be effective for aspergilloma, even in patients with massive hemoptysis.4,8 This modality has been recommended for cases of recurrence of life-threatening hemoptysis after BAE.
Itraconazole is an orally administered antifungal agent with activity against A. fumigatus and high tissue penetration into the lung. The use of itraconazole for aspergilloma has been reported in several noncontrolled studies.4,8 Data from these studies showed that the use of itraconazole capsules at doses ranging from 200 to 400 mg/d for 6 to 18 months resulted in radiographic and symptomatic improvement in almost two-thirds of the patients and may have a place for the treatment of aspergilloma.8 Serum itraconazole levels were not measured in most of these studies, but a recent study of treatment of pulmonary aspergilloma with itraconazole (100–200 mg/d) demonstrated sufficient itraconazole levels within the aspergilloma cavities.4,8 The major limitation of itraconazole is that it works slowly; thus, use of it would not be prudent in cases of life-threatening hemoptysis. In addition, recurrence of aspergilloma often follows discontinuation of itraconazole treatment, whereas acquisition of secondary resistance of A. fumigatus isolates to itraconazole has been described in patients with aspergilloma following prolonged treatment. Voriconazole has been successfully used to treat pulmonary aspergilloma caused by itraconazole-resistant A. fumigatus isolates, although cases of voriconazole failure in patients with aspergillomas have also been reported. There is evidence of development of secondary resistance to voriconazole during long-term treatment of patients with aspergillomas.
Invasive Bronchial Aspergillosis
The term invasive bronchial aspergillosis (IBA) refers to infection involving the large airways. Further subclassification of IBA based on the bronchoscopic appearance has been proposed, including tracheobronchitis, pseudomembranous tracheobronchitis, and ulcerative tracheobronchitis. In fact, these entities represent a continuous spectrum of this infection.7,8 An alternative, more functional classification based on the depth of tissue invasion by Aspergillus includes (a) superficial infiltration type; (b) full-layer involvement type; (c) occlusion type (airway occlusion ≥50% of the original diameter of involved bronchi; and (d) mixed type. The potential impact of this classification system on the management of IBA has not been validated.
Aspergillus tracheobronchitis is the least invasive form of IBA and is characterized by the presence of a superficial tracheobronchial inflammation, intact mucosa with no evidence of ulcers, and pseudomembranes or other abnormalities; Aspergillus spp. are identified in mucus exudates.7,8 In contrast, pseudomembranous tracheobronchitis is characterized by significant necrosis of bronchial epithelium and formation of pseudomembranous plaques, which are white, gray, or black in color. These lesions may progress and result in extensive invasion of the large airways; however, in the majority of patients, the depth of invasion does not extend beyond the bronchial cartilage.7,8 Finally, ulcerative tracheobronchitis is the most aggressive form of IBA and manifests with endobronchial plaques, nodules, or areas of ulceration and necrosis; in addition, the infection may extend to the adjacent pulmonary parenchyma and pulmonary vasculature.7,8 Late sequelae of ulcerative tracheobronchitis include the formation of excessive granulomatous tissue with resulting bronchial stenosis.
The pseudomembranous form of IBA is typically seen in lung transplant recipients, who accounted for almost 40% of published cases.6,8 In fact, IBA is the most common infection within 3 months following lung transplantation, whereas IPA tends to occur much later in this patient population. Occasionally, pseudomembranous tracheobronchitis is found in various clinical settings including HSCT, hematological malignancies, metastatic renal cell carcinoma, post-influenza, COPD, AIDS, systemic lupus erythematosus, metabolic abnormalities including DM, and even in immunocompetent individuals.7,8 Patients may be initially asymptomatic or have symptoms attributed to the underlying illness with negative chest radiographs. However, as the infection progresses, symptoms become more pronounced, including characteristic stridor, resulting in respiratory failure and ultimately death. Persistent stridor in neutropenic or severely immunocompromised patients should raise suspicion for IBA; however this symptom occurs in less than one-quarter of all cases. Importantly, Aspergillus galactomannan antigen has limited value in IBA diagnosis as it is positive in only 60% of the cases.5,6
The ulcerative form of IBA occurs almost exclusively at the site of bronchial anastomosis in lung transplant recipients.6 Bronchoscopically, the appearance is that of severe tracheobronchitis with multiple ulcers at the site of anastomosis. The symptoms are similar to those of pseudomembranous IBA. Complications include acute IPA, severe bronchial stenosis, anastomotic dehiscence, and bronchial necrosis with bronchoarterial anastomotic fistula.6 Rarely, ulcerative IBA develops in patients with AIDS or solid tumors.6 Diagnosis of all forms of IBA requires a high index of suspicion and is established only by bronchoscopic examination.
Bronchial stump aspergillosis is an unusual complication of lung resection.5–7 The period from pneumonectomy to the onset of infection usually ranges from 6 to 12 months, although noted to occur after 3 years in one case. Patients may present with a productive cough and hemoptysis, sputum that may be putrid, and occasionally, expectoration of fungal material or suture thread.5–7 Chest radiographs are usually unchanged when compared with those taken at baseline. The cause is secondary Aspergillus infection of silk suture material used to close the bronchus after pulmonary resection. Local inflammation, compromised tissue viability, and the use of silk sutures favor the establishment of Aspergillus infection. Substitution of nylon monofilament for silk sutures at surgery has significantly reduced this infectious complication.5–7
All of the forms of IBA, including bronchial stump anastomosis, require prompt treatment with systemic antifungals for IA (discussed in detail below). Data from controlled studies of the treatment of IBA are lacking, however. A common, although unproven practice is to combine systemic antifungal therapy, such as intravenous AMB-D or voriconazole, with aerosolized AMB-D.5–7 Notably, most reported failures occurred following cessation of the treatment. Finally, surgical resection and stent placement may be necessary in conjunction with antifungal therapy if dehiscence of the anastomosis occurs because of tracheobronchial aspergillosis.5–7
Chronic Pulmonary Aspergillosis
Chronic forms of Aspergillus infection of the lung have long been recognized.8 CPA occurs in patients with chronic cavitary lung disease and is characterized by an indolent clinical course evolving over months to years, constitutional symptoms, serum precipitins to A. fumigatus, elevated acute phase markers of inflammation, and an immune status that ranges from normal to mild immunosuppression. Locally invasive (semi-invasive) Aspergillus infection may be evident histopathologically. However, both angioinvasion and dissemination are absent. A variety of terms have been used for CPA, including pulmonary aspergillosis with cavitation, complex aspergilloma, chronic granulomatous aspergillosis, semi-invasive pulmonary aspergillosis, and CNPA.8
In an attempt to better define CPA, a subclassification of this entity based on clinical and radiographic findings was recently proposed, introducing the terms CNPA, chronic cavitary pulmonary aspergillosis (CCPA), and chronic fibrotic pulmonary aspergillosis (CFPA).8 CNPA comprises a syndrome of slowly progressive cavitary lung disease, chronic respiratory symptoms, and the presence of precipitating antibodies against Aspergillus. Some cases of CNPA have invasion of the lung parenchyma by Aspergillus spp. However, in most of the described cases, there is no tissue invasion despite the presence of extensive and progressive tissue damage. CCPA refers to cases in which there is formation and expansion of multiple cavities over time, whereas CFPA refers to cases in which cavity formation is followed by a pronounced fibrotic reaction.8 In addition, it has been recommended that any case with proven hyphal invasion of tissue should be classified as CNPA.
Defects in mucociliary clearance associated with structural lung disease appear to be a critical factor in the pathogenesis of CPA.8 Prior mycobacterial lung infection, emphysema and/or COPD, bullae, asthma, sarcoidosis, pneumoconiosis, lung cancer, thoracic surgery, upper lobe fibrosis complicating ankylosing spondylitis, marijuana use, and a history of Legionella infection have been described as predisposing conditions for CPA. In addition, subtle but essential defects in innate immunity may play a role in the pathogenesis of CPA (see Chapter 121). Hence, defects in mannose-binding lectins, alterations in SP-D, certain polymorphisms in cytokine encoding genes such as transforming growth factor-β, TNF-α, IFNγ, and IL-15, and systemic illnesses associated with a degree of immunosuppression, such as corticosteroid-based therapy, diabetes mellitus, AIDS, and alcohol abuse, have all been associated with CPA in humans.8 However, the precise pathophysiological mechanism of new cavity formation remains uncertain.
CPA tends to affect middle-aged individuals who are relatively immunocompetent and more male than female individuals.8 CPA has an indolent and progressive course that lasts for years. Chronic productive cough and weight loss with mild hemoptysis, dyspnea, and fatigue are the usual presenting symptoms. Pleural fibrosis and Aspergillus empyema appear to complicate some cases of CPA.8
Although the diagnosis of CPA may be suspected with a single chest radiograph, sequential chest radiographs are typically required to confirm the progressive nature of CPA lesions. CT is particularly useful in defining the precise pattern and extent of the disease. Typical radiographic findings include the presence of one or more cavities, which may or may not contain fungus balls, often located in the upper lobes.8 New cavity formation and expansion of pre-existing cavities are also characteristic of CPA. Pericavitary infiltrates and adjacent pleural thickening are frequently observed and appear to correlate with the overall disease activity.8 With appropriate treatment, these radiographic abnormalities may regress, leaving residual thin-walled empty cavities.
The combination of characteristic clinical and radiological findings and either the presence of Aspergillus precipitins or the isolation of Aspergillus spp. from respiratory samples is highly indicative of CPA.8 Although the demonstration of precipitating antibodies to Aspergillus is the cornerstone and a prerequisite for the diagnosis of CPA, these antibodies may be negative, especially in case of infection with non-fumigatus Aspergillus spp.8 Histopathology demonstrates chronic inflammation and fibrosis, sometimes with granulomatous features with or without visible hyphae. The absence of hyphae does not exclude the diagnosis of CPA because of the paucity of high fungal burden in patients with CPA.
Differentiating CPA from other serious lung conditions that require specific therapy, such as cancer and chronic cavitary mycobacterial and endemic fungal infections, as well as other inflammatory lung conditions is a major challenge as overlap is not uncommon. In fact, there is a strong association between CPA and atypical mycobacterial infections. In these cases, imaging is not helpful, so biopsy and comprehensive microbiological studies must be used to exclude the co-existence of CPA with such entities. In addition, CPA differs from simple aspergilloma because of the presence of constitutional symptoms, the development of persistent pericystic lung nodules or pleural thickening, consolidations or ground-glass opacities, and the development and/or progression of cavities.8 Finally, one should always consider the possibility of a pyogenic infection in a cavity that may require drainage and appropriate antibacterial treatment.
CPA requires prolonged treatment with systemic antifungal agents. In the literature, most patients with CPA have received treatment with oral itraconazole, whereas IV AMB-D has been successfully used in refractory cases.8 In a series of 18 patients, use of itraconazole capsules resulted in improvement or stabilization of disease in 71% of the cases.8 Use of voriconazole has been associated with favorable responses in small case series with response rates ranging from 30% to 80% and should be considered as another treatment option. Posaconazole has also successfully used with 61% response rate in 79 patients with CPA. However, emerging rates of azole resistance in Aspergillus isolates recovered from patients with CPA have been reported, although there is controversy on whether in vitro resistance of A. fumigatus to azoles correlates with worse clinical outcome. In addition, serious side effects necessitating discontinuation of treatment with azoles occur in up to 27% of the patients. Alternative treatment strategies include intravenous use of polyenes or echinocandins. Although the reported response to IV AMB-D (81%) appears to be slightly better than that to itraconazole, administration of AMB-D may require prolonged hospitalization and is associated with considerable toxicity, especially nephrotoxicity.8 In a recent randomized study, treatment with IV micafungin had comparable (60%) rates of response with IV voriconazole (51%) and significantly less adverse effects in patients with CPA and could represent an alternative especially in cases where azole resistance in a concern.6 Anecdotal reports of improved outcome as a result of adjunctive immunomodulating treatment with IFN-γ have also been reported.8
Surgery plays a limited role in the treatment of CPA because of the poor overall lung function in many patients. Major postoperative complications, such as respiratory failure, bronchopleural fistulae, pleural extension of aspergillosis, and even dissemination of Aspergillus can occur.8 Thus, surgery should be reserved for selected patients with a reasonable respiratory reserve for whom there are no other treatment options. For example, surgery may be appropriate for patients with severe hemoptysis if embolization fails.
Although systemic antifungal therapy for CPA seems to be beneficial, assessment of response to it is difficult to gauge because the activity of the disease may fluctuate over time. Weight gain and improved energy levels are the earliest and most reliable indicators of response. Inflammatory markers also improve albeit more slowly, and they usually remain elevated even during long-term therapy. Relapse months or years after discontinuation of treatment is frequently reported.
Invasive Pulmonary Aspergillosis
An important clinical manifestation of Aspergillus-related disease is invasive pulmonary aspergillosis, discussed in detail below.
Epidemiology Aspergillus has emerged as one of the most common causes of infectious death in severely immunocompromised patients, with mortality rates approaching 70% in patients with leukemia and recipients of HSCTs.2,5–7 Surveillance studies demonstrated a three- to fourfold increase in the frequency of Aspergillus infections at major cancer centers over the past two decades. In fact, IPA has surpassed invasive candidiasis as the most common fungal infection found at autopsy at several institutions; approximately 15% to 20% of patients with leukemia die of fungal pneumonia caused by Aspergillus spp. Similarly, in allogeneic HSCT recipients, 15% to 30% of deaths are caused by refractory fungal infections, mainly caused by Aspergillus spp., and most of these infections occur late in the postengraftment period in the setting of graft-versus-host disease (GvHD).2,5–7 Large prospective multicenter surveillance studies contacted by the Transplant-Associated Infection Surveillance Network (TRANSNET) and Prospective Antifungal Therapy (PATH) Alliance has furthered insights into the epidemiology of IA in the United States. From both studies, IA has emerged as the most common invasive fungal infection in HSCT and solid-organ transplant recipients (43% and 59% of all IFIs, respectively) over invasive candidiasis and mucormycosis, with a 12-month cumulative incidence of 1.6%. Overall mortality in these patients was estimated to be approximately 36% and 75% at 12 weeks and at 1 year, respectively.5–7
Changes in HSCT practices over the past decade have greatly impacted the epidemiology of IPA. Autologous HSCT is associated with a low risk of IPA because of shortened periods of preengraftment neutropenia and a lack of GvHD. On the other hand, IPA has a bimodal distribution in allogeneic HSCTs, with patients at risk before and after bone marrow engraftment. The increasing use of peripheral blood stem cells, growth factors, and nonmyeloablative conditioning regimens (mini-transplants) in allogeneic HSCT has shortened the duration of preengraftment neutropenia and the incidence of IPA shortly post HSCT. In contrast, late (40–80 days after engraftment) and very late (80+ days after engraftment) IPA has increased.5–7 This trend is associated with increased use of matched unrelated-donor (MUD) allogeneic transplants and with interrelated risk factors, such as lymphopenia, chronic GvHD, corticosteroid-based therapy, and/or CMV infection.5–7 Among HSCT recipients, IPA now predominantly occurs as a community-acquired rather as a nosocomial infection in nonneutropenic patients late after engraftment, in whom GvHD and management with increasingly intense immunosuppression have emerged as the major risk factors. Of interest, recent studies in HSCT recipients implicate geoclimatic changes, associated with high environmental spore counts following periods of high temperature and low precipitation, as a factor for the development of IPA.
In addition to the “classic” groups of patients at risk for IPA, such as those with prolonged, profound neutropenia because of a hematological malignancy (5%–25% risk) or aplastic anemia; recipients of allogeneic HSCTs (5%–30% risk), or lung transplants (17%–26% risk); those with AIDS, severe combined immunodeficiency, or CGD (25%–40% lifetime risk); burn patients; and patients receiving chronic corticosteroids, IPA has been increasingly described in new groups of patients.5–7 Hence, patients with systemic lupus erythematosus, patients with multiple myeloma who receive high-dose steroids, and premature neonates are prone to the development of severe IPA. Furthermore, several cases of IPA have been lately reported in patients with Crohn disease or rheumatoid arthritis who received treatment with TNF inhibitors (e.g., infliximab).5–7 Even the use of high-potency inhaled steroids may predispose some apparently normal hosts to IPA in rare instances. IA is considered an emerging infection in patients with prolonged stay in intensive care units (ICUs). ICU aspergillosis is a distinct clinical entity that is probably underreported.2,5–7
IPA caused by Aspergillus spp. less susceptible to antifungals than Aspergillus fumigatus such as A. terreus and A. flavus has also been reported in individual institutions.2,5–7 This phenomenon may be at least partially a reflection of antifungal selection pressure determined by extensive empirical and prophylactic use of antifungal agents in high-risk patients. Alternatively, because non-fumigatus aspergilli cause nosocomial infections more frequently than does A. fumigatus, an epidemiological niche for these species may also account for their predominance in some institutions. A. terreus is clinically resistant to AMB, but species, including A. flavus, Aspergillus lentulus, Aspergillus nidulans, Aspergillus ustus, Aspergillus glaucus, and others, can also demonstrate resistance to antifungal agents.
A number of nosocomial clusters of IPA have been reported over the past three decades mostly associated with hospital construction and defects in air-handling equipment. Air and environmental surfaces have been the focus of most investigations. Water has been implicated as an additional source of nosocomial transmission of Aspergillus spp., although this has not been confirmed in other studies.2,5–7 An outbreak of IPA after major cardiac surgery caused by abnormally high levels of fungal spores in the air of the ICU has been recently reported.
Pathogenetic Mechanisms Because Aspergillus is a saprophytic mold that colonizes in the respiratory tract of immunocompromised patients, the pathogenesis of infection depends on the host’s underlying immunosuppression. In the immunocompromised host, IPA typically occurs following inhalation of Aspergillus conidia, although hematogenous dissemination from a cutaneous or gastrointestinal source is seen occasionally.1,2 Damage to respiratory epithelium because of radiation therapy, chemotherapy, GvHD, or prior infection (e.g., RSV, influenza) may facilitate attachment of Aspergillus conidia to the respiratory epithelial surface.5
Breach of mucosal host defense mechanisms in the setting of underlying immunodeficiency places the host at risk for invasive infection by Aspergillus spp. Thus, once Aspergillus has invaded the respiratory tract, control of the infection is highly dependent on functional adaptive immune responses. Lymphopenia and local/global dysregulation of the host adaptive Th1/Th2 polarization have been shown to play a key role in the subacute progression of IPA in nonneutropenic hosts.5–7 Hence, in the late postengraftment period, allogeneic HSCT recipients have a low IFN-γ/IL-4 ratio, which is suggestive of a Th2 response that potentially accounts for their susceptibility to IPA.3–5,7 In comparison, healthy individuals mount a predominantly Th1-type cellular response to Aspergillus infection. In humans, iatrogenic suppression of protective Th1 responses is common, particularly in the setting of chronic GvHD (an excessive Th1 response of allograft T lymphocytes) treated with high-dose corticosteroids and/or other immunosuppressive regimens.3,5,7 Importantly, Aspergillus-mediated suppression of host Th1 responses by production of gliotoxin is also plausible.2,3 Similarly, recent preclinical data indicate that herpes virus reactivation and bacterial co-infection in the lungs induce profound dysregulation of immune responses, facilitating the development of IPA.2,3,5–7 For example, CMV infection can result in dysfunction of dendritic cells and phagocytes, which may predispose patients to IPA. Of interest, critically ill patients with prolonged ICU stay develop a systemic deactivation of the immune system following sepsis or any severe insult that has activated a systemic inflammatory response.3 This sepsis-induced immune paralysis often results in superinfection with opportunistic pathogens, including Aspergillus. In agreement, mice recovering from sepsis remain exquisitely susceptible to Aspergillus and develop IPA upon pulmonary infection without the need for immunosupression.3 Although the mechanism of increased susceptibility to Aspergillus in sepsis-induced immunosupression remains obscure, there is evidence of predominance of a deregulated myeloid cell response with increased levels of IL-13, IL-4, TGF-β in the lung of mice that develop invasive fungal infection. Importantly, deregulated cytokine release by myeloid cells and associated susceptibility to IPA in septic mice could be restored by adoptive transfer of bone marrow–derived dendritic cells from healthy mice.3
Once germination of conidia occurs, Aspergillus hyphae invade pulmonary arterioles and lung parenchyma, leading to ischemic necrosis. Hematogenous dissemination with thrombosis, hemorrhagic infarction, and invasion of distant organs may result from invasion of arterioles by Aspergillus hyphae and is found in approximately one-third of cases of IPA at autopsy.2,5–7 Thus, IA, in addition to pneumonia, can present with a clinical picture similar to that of other thrombotic and embolic diseases, such as pulmonary embolism, cerebral vascular accidents, Budd–Chiari syndrome, and renal papillary necrosis. In addition, IA can spread to contiguous structures, across the diaphragm to the stomach, or from the lung to the heart or superior vena cava.
Importantly, it has been recently demonstrated that IPA has different pathophysiological mechanisms depending on the type of immunosuppression. Thus, a low Aspergillus tissue burden, extensive inflammatory injury, absence of angioinvasion, and localized infection have been observed in corticosteroid-immunosuppressed mice, whereas minimal inflammation, extensive angioinvasive Aspergillus growth, and disseminated infection are characteristic of IPA in cyclophosphamide-treated neutropenic mice.2,5–7 In corticosteroid-treated mice, host immune effector cells appear to have limited activity against Aspergillus hyphae but cause extensive alveolar damage and exudative bronchiolitis, which appears to be more important than the Aspergillus-induced injury itself. This nonangioinvasive form of IPA has been increasingly recognized in a wide range of nonneutropenic hosts, including those who received corticosteroids, recipients of solid-organ transplants and allogeneic HSCTs with GvHD, patients with AIDS, and patients with CGD (Fig. 133-6).2,5,7 Similar to the animal models of IPA, histopathology in those nonneutropenic patients is characterized by extensive pyogranulomatous inflammatory reactions, inflammatory necrosis, and extensive cavitation.
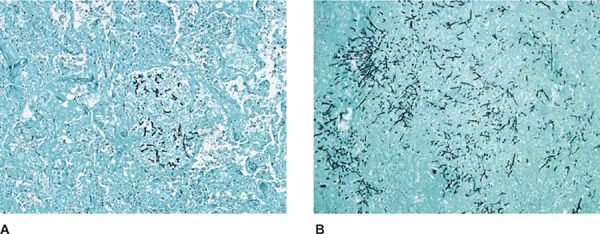
Figure 133-6 The characteristic pattern of Aspergillus invasion in tissue obtained from (A) an HSCT recipient with IPA and severe GvHD and (B) a patient with acute leukemia and severe neutropenia in whom IPA developed Extensive inflammation, less Aspergillus burden and absence of angioinvasion was observed in the patient with GvHD. In comparison, the histopathology of IPA in the patient with severe neutropenia was characteristic for scant infiltration by phagocytic cells, extensive coagulative tissue necrosis caused by angioinvasion, and a high fungal burden. Gomori methenamine silver stain, ×100. (Used with permission of Dr. M. Luna.)
Although deficiency of TLR adaptive protein MyD88 and Dectin-1 in humans is not associated with the development of IA, genetic polymorphisms in TLR-1, TLR4, TLR-6, and Dectin-1 predispose to the development of IA in allogenic HSCT recipients.3 In addition, polymorphisms in IL-10 and plasminogen are associated with increased risk for IA. Thus, better understanding immunogenetics of IA in these high-risk individuals could facilitate risk stratification and design of preventive strategies.3
Clinical Features Symptoms of IPA begin with fever (unless the patient is receiving corticosteroids), which may be followed by a mild nonproductive cough suggestive of bronchitis. Pleuritic chest pain and progression to pneumonia occur within 1 to 2 days. Cavitation tends to occur in patients if immunosuppression is decreased, such as in leukemic patients during recovery of bone marrow hematopoiesis or when steroid-based therapy is reduced significantly, and on occasion gives rise to massive hemoptysis (Figs. 133-7 and 133-8).2,5–7 Pleuritic pain and slight hemoptysis may suggest pulmonary infarction. Patients also may expectorate necrotic tissue filled with hyphae. Cough, sputum production, and pleural effusion are either absent or minimal. IA must be strongly considered when dealing with susceptible patients in whom treatment with broad-spectrum antibiotics fails.
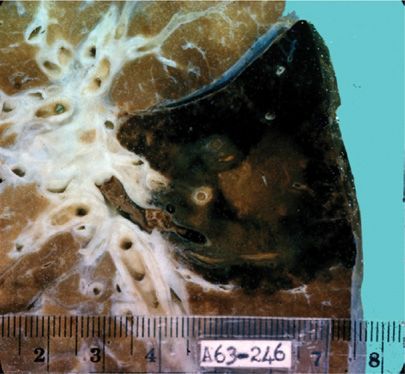
Figure 133-7 A 62-year-old man with acute leukemia in whom IPA developed. The gross appearance of the fungal lesion shows a central necrotic area surrounded by a lining of hemorrhagic infarction. (Used with permission of Dr. G.P. Bodey.)
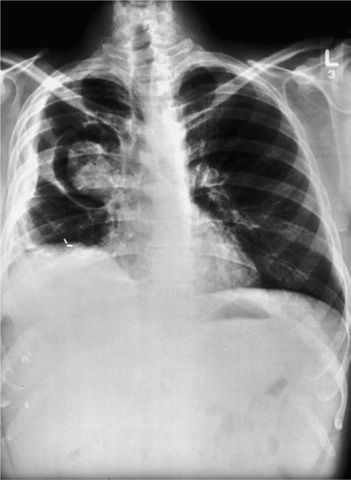
Figure 133-8 The characteristic radiographic features of IPA following neutrophil recovery illustrated in a patient with acute leukemia. A lesion has cavitated, creating the air crescent sign. (Used with permission of Dr. G.P. Bodey.)
Extrapulmonary sites may be involved including the highly vascular organs such as the kidneys, liver, spleen, and central nervous system. Invasive disease of the nose and paranasal sinuses occurs in immunocompromised patients, and contiguous invasive spread into the orbit or into the cranial vault can result in a syndrome similar to rhinocerebral mucormycosis.
Diagnosis Substantial delay in establishing an early diagnosis remains a major impediment to the successful treatment of IPA. Chest radiographs are not sensitive in detecting early forms of bronchopulmonary IPA, and up to 10% of patients with proven IPA have “normal” chest radiographs within a week of death.2,5–7 High-resolution CT scans of the chest may reveal small wedge-shaped subpleural lesions or nodules typically surrounded by intermediate attenuation. This halo sign correlates with hemorrhage and edema surrounding an infarct caused by thrombosis and is highly suggestive of acute IPA, especially in neutropenic patients with leukemia.2,5–7 However, the halo sign has been documented only in 33% to 60% of patients and is transient. In fact, to be useful for the diagnosis of IA, CT must be performed within 5 days of the onset of infection, because more than 75% of initial halo signs disappear within a week.2,5–7 With neutrophil recovery, these lesions coalesce and cavitate, forming the “air crescent” sign, a classic sign of late filamentous invasive mold infection (IMI). However, the air crescent sign does not appear until the third week of the infection, and the appearance may be too delayed to be helpful in the diagnosis of IA. Importantly, lesions frequently increase in size in the first 7 to 10 days following effective antifungal therapy. In view of the angioinvasive nature of IPA, detection of vascular occlusion by CT-based pulmonary angiography was evaluated in a recent study as an early sign of IMI. Importantly, the presence of positive vascular occlusion sign was more sensitive and specific than the halo sign and comparable to serum galactomannan test for diagnosis of IPA. Thus, CT pulmonary angiography could represent a viable diagnostic tool for IPA, especially in patients at low-risk of contrast-induced acute kidney injury.
Persistently febrile neutropenic patients with leukemia would benefit from the use of early high-resolution chest CT to identify early radiologic signs of IPA such as the halo sign. In a recent study of febrile neutropenic patients, early and routine CT scanning coupled with early intensive antifungal-based treatment and surgery resulted in increased survival rates when compared with therapy initiated at the first sign of a pulmonary fungal infection.2,5 CT also plays a critical role in determining the appropriateness of various downstream diagnostic procedures (bronchoalveolar lavage [BAL], percutaneous needle biopsy, and open lung biopsy) in patients with IPA. Nonetheless, clinical and radiologic presentations of IPA in the expanding group of nonneutropenic patients who develop infection is atypical, including cases with tree-in-bud opacities or diffuse ground-glass appearance in CT.2,5–7
Respiratory cultures of Aspergillus from expectorated sputum, bronchial washings, or BAL specimens have low sensitivity (<30%) for diagnosing IA but a high positive predictive value (>60%) in heavily immunocompromised patients.5 Early performance of CT-guided fiberoptic bronchoscopy with BAL in immunocompromised patients with pulmonary infiltrates has been shown to improve the diagnostic yield of this procedure. In addition, there is a need for the development of standardized BAL protocols in order to facilitate comparative clinical studies on the diagnosis of IPA. Blood cultures have little diagnostic value for IA but may reflect true disease cases of A. terreus infection. Ultimately, open lung biopsy may be required for a definitive diagnosis of IPA and may not be feasible until late in the course of infection or recovery of pancytopenia.
In recent years, efforts have been directed toward identifying non–culture-based markers for rapid, reliable diagnosis of IA.5–7 Those based on the detection of anti-Aspergillus antibodies have very poor sensitivity for IA, especially in patients receiving immunosuppressive agents. Instead, tests based on identifying circulating fungal antigens or metabolites are promising. To date, the two approaches that have demonstrated the greatest promise in early clinical studies are detection of antigens and measurement of Aspergillus nucleic acids by using PCR.
Galactomannan is a polysaccharide cell wall component of Aspergillus spp. that is released into the circulation during fungal growth in tissues and can be detected in the serum of patients with IA.5–7 A sandwich enzyme–linked immunosorbent assay (ELISA) capable of detecting galactomannan at concentrations as low as 0.5 ng/mL has been recently developed. Initial studies evaluating the role of galactomannan serum assay in the diagnosis of IA have been predominantly conducted with profoundly neutropenic patients undergoing chemotherapy for cancer or recipients of HSCTs who were on fluconazole prophylaxis. These studies have documented sensitivity rates ranging from 67% to 100% and specificity rates ranging from 86% to 99% in these patients.5–7 When serially monitored, the galactomannan assay preceded the diagnosis of IA by an average of 6 to 14 days. However, in a recent study that evaluated the value of the galactomannan assay in the detection of early IA, the appearance of major lesions (halo sign, air crescent sign, and cavitation) on high-resolution CT scans almost coincided or even preceded the detection of the galactomannan antigen in serum.5–7 Furthermore, uncertainties remain regarding the performance of this assay in other settings, such as breakthrough IA to mold-active antifungal prophylaxis, pediatric populations, and solid-organ transplant recipients. Other factors, such as the pretest probability of infection, the patient’s immune status (neutropenia vs. GvHD), antifungal therapy, antibacterials, and diet may affect both the performance and interpretation of the galactomannan assay. Hence, sensitivity rates as low as 30% have been reported in nonneutropenic HSCT recipients receiving mold-active antifungal prophylaxis and lung transplant recipients.5–7 In addition, false-positive results have been described in patients receiving β-lactam antibiotics, particularly piperacillin–tazobactam, as well as in those ingesting certain cereals, pastas, nutritional supplements, and soy sauce produced with fermentation products of Aspergillus oryzae, a fungus commonly used in food production.5,7 In addition, false-positive galactomannan antigenemia has also been reported as a result of transfusion of blood products. Recent studies evaluated the performance of detection of galactomannan in other fluids, particularly BAL and showed that BAL GM has improved sensitivity compared to serum GM in high-risk patients with IPA.2,5–7 A recent meta-analysis of 30 diagnostic studies with the use of BAL GM, using an otimal cutoff value of 1.0, the BAL-GM assay had 90% sensitivity and performed better compared to PCR and serum GM test although specificity was lower (89%).
A colorimetric assay for the detection of 1,3-β-D-glucan, an integral cell wall component in a number of pathogenic yeasts and filamentous fungi, recently became commercially available.5,7 The sensitivity and specificity rates for this test in limited studies thus far have ranged from 67% to 100% and from 84 to 100%, respectively. Several false-positive test results have been reported in patients with cirrhosis, in patients undergoing hemodialysis, in patients following abdominal surgery, and in those receiving chemotherapy with particular agents.5,7 Furthermore, the 1,3-β-D-glucan assay has been reported to be less sensitive and reproducible and become positive later in the course of IA when compared with galactomannan antigen assay.
PCR detection of Aspergillus nucleic acids is a promising method for early detection of IA. The sensitivity of PCR is excellent, but its specificity for invasive infections can be problematic, and false-positive results are common.5,7 Multiple unresolved issues accompany the use of PCR for the diagnosis of IA, including the sample type (serum, BAL fluid), amplification strategy (nested vs. conventional PCR), protocol (real-time quantitative vs. conventional PCR), and primer selection (Aspergillus-specific or pan-mold, pan-fungal primers). There have been no good studies examining how PCR performs in comparison with galactomannan detection for the early diagnosis of IA. However, it has been shown that PCR is inferior to serum galactomannan assay in animal models and in some but not all human studies.5–7 A recent meta-analysis of 19 studies comparing the value of BAL PCR with BAL GM concluded that although sensitivity and specificity of both tests were comparable, combination of both tests increased sensitivity without compromising specificity. Thus, like GM testing, PCR is likely to become an adjunctive diagnostic method alone or in combination with other biomarkers and standard microbiological, histological, and radiographic methods of diagnosing IA.
Novel investigational diagnostic approaches include the development of a breath test to detect volatile Aspergillus biomarkers that are not produced by other pathogens, and detection of circulating Aspergillus antigens in BAL and serum of patients with IPA by using an immunochromatographic lateral flow device.
Treatment of IPA Although the past two decades have witnessed improved outcomes of patients with IA, the mortality rate of this mycosis in severely immunocompromised patients remains unacceptably high. Several strategies have been applied in an attempt to improve the outcome of IA. These include the introduction of potent antifungal agents such as voriconazole, early onset of treatment triggered by CT and/or galactomannan antigen, use of delivery systems to improve the therapeutic index of currently available antifungals such as lipid formulations of AMB, and possibly combination antifungal therapy, surgical excision of sequestered necrotic lesions, and use of immunomodulating agents in selected patients.6 There has not been a systemic evaluation of the merit of each of these strategies in the treatment of IA, however. This is not surprising in view of the complexity of IA and the paucity of organized, prospective controlled clinical studies in refractory IA.
Tables 133-5 and 133-6 list the principles of management of IPA. Until early 1990 s, AMB-D was the only drug available for the treatment of aspergillosis, and no prospective evaluations of this drug had been attempted.5–7 Another major obstacle to evaluating the merit of antifungal therapy for IPA until the early 2000s was difficulty in establishing the diagnosis; hence, most patients received empirical treatment. The introduction of uniform diagnostic criteria for proven, probable, and possible IA by the European Organization for Research and Treatment of Cancer (EORTC)/Mycoses Study Group (MSG) consortium has been an advance.9 These criteria are standardized and reproducible and consist of host-risk, microbiological, clinical/radiological, PCR, and/or galactomannan antigen criteria. Despite the fact that the EORTC/MSG criteria have been updated to incorporate biomarkers for the diagnosis of IMI, their clinical applicability was questioned in real word setting; for example, in an autopsy study more than 60% of proven cases of IA in patients with hematological malignancies were not identified antemortem according standard definitions.10 In addition, there is lack of a standardized PCR protocol for the detection of Aspergillus DNA from clinical specimens with validated, high intralaboratory quality performance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


