Chapter 8
Arteriogenesis and Angiogenesis
Kathleen M. Lamb, Paul J. Dimuzio
Based on a chapter in the seventh edition by Paula K. Shireman and Carlo O. Martinez
Peripheral arterial disease (PAD) of the limbs can progress to critical limb ischemia (CLI), characterized by rest pain and tissue loss, including nonhealing ulceration and gangrene.1,2 The body compensates via neovascularization, through which it forms collateral circulation to bypass the obstructed vessel (arteriogenesis) or increases capillary density (angiogenesis) to deliver oxygen and nutrients to ischemic tissue. Despite these responses, disease progression can lead to amputation, decreased quality of life, comorbidity, and death, and is an economic burden to the health care system.
Traditional treatment of PAD includes risk factor modification (tobacco abuse, diabetes, hypertension, hyperlipidemia), exercise programs, and medical therapy (antiplatelet agents, phosphodiesterase inhibitors).1–3 As atherosclerosis progresses, more invasive intervention may be necessary, including endovascular therapies and surgical bypass. Patients with CLI may not be candidates for surgical intervention because of severe medical morbidity or nonreconstructable disease. This patient population may be candidates for biologic treatments, including gene-based, molecular, and cell-based therapies designed to promote healing and prevent amputation.
Advances in basic science research have developed these biologic therapies over the past 2 decades. Early clinical trials focusing on gene and molecular therapies, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), have demonstrated limited benefit. More promising results have been observed using cell-based therapies, including endothelial progenitor cells (EPCs) and bone marrow–derived mononuclear cells (BM-MNCs).
Herein, the basic science and processes involved in neovascularization (specifically arteriogenesis and angiogenesis) as well as recent human clinical trials designed to promote neovascularization for CLI are discussed.
Neovascularization
Neovascularization refers to the formation of new blood vessels and includes vasculogenesis, arteriogenesis, and angiogenesis.4
Vasculogenesis is the de novo formation of embryonic blood vessels from vascular progenitor cells or hemangioblasts, which develop into hematopoietic precursors and endothelial cells.5 These cells induce differentiation of discrete vascular layers as cells transform into endothelial cells, smooth muscle cells (SMCs), and adventitial pericytes. Although vasculogenesis primarily occurs during the embryonic stages of development, postembryonic vasculogenesis results from either arteriogenesis or angiogenesis.6,7
Arteriogenesis involves growth of collateral vessels and remodeling from preexisting arterial-arteriolar connections.6,8 Arteriogenesis is induced by a change in hemodynamic forces (fluid shear stress [FSS]) resulting from pressure differences within a diseased artery. Fluid shear stress activates the endothelium, leading to increased transcription of the promoter regions of a variety of proteins contributing to vessel growth4 (Table 8-1). With progressive arterial stenosis, blood follows from the path of least resistance and is shunted into collateral vessels.4 Collateral vessel formation maintains flow beyond the obstruction, hopefully sufficient enough to maintain distal tissue health.
Table 8-1
Transcription and Growth Factors Influencing Arteriogenesis and Angiogenesis
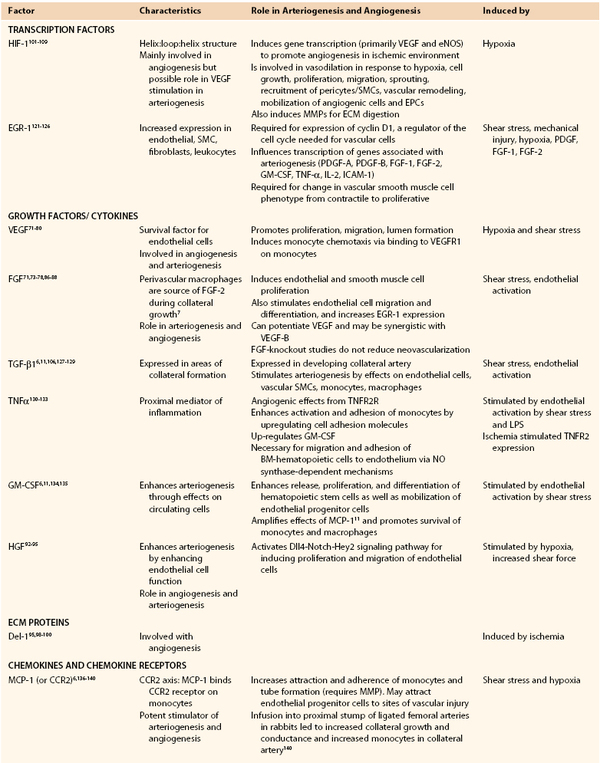
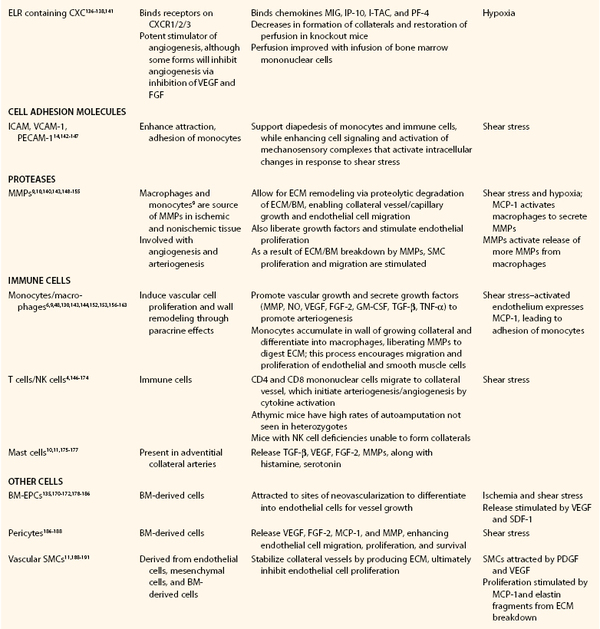
BM, Bone marrow; BM-EPCs, bone marrow–derived endothelial progenitor cell; EGR-1, early growth response protein-1; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; FGF, fibroblast growth factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HGF, hepatocyte growth factor; HIF-1, hypoxia-inducible factor-1; IL, interleukin; ICAM, intracellular adhesion molecule; MCP-1, monocyte chemotactic protein-1; MIG, monokine inducible gamma-interferon; IP-10, interferon gamma-induced protein 10; I-TAC, interferon-inducible T-cell alpha chemoattractant; PF-4, platelet factor 4; MMP, matrix metalloproteinase; NK, natural killer; NO, nitric oxide; PDGF, platelet-derived growth factor; PECAM, platelet endothelial cell adhesion molecule; SDF-1, stromal cell–derived factor-1; SMC, smooth muscle cell; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α; TNFR2R, tumor necrosis factor receptor 2R; VCAM, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
Adapted from Huang P, et al: Autologous transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells improves critical limb ischemia in diabetes. Diabetes Care 28:2155-2160, 2005.
Angiogenesis, which refers to the sprouting of new capillaries from preexisting ones, is stimulated by decreases in oxygen tension secondary to reduced tissue perfusion. Local ischemia stimulates an increase in hypoxia-inducible factor-1 (HIF-1), which results in increased production of VEGF, a potent angiogenic factor. VEGF induces endothelial cell proliferation and increases endothelial permeability.8 Matrix metalloproteinases (MMPs) locally degrade basement membrane and extracellular matrix, allowing vessel growth and increase in tissue perfusion.9–11 Although this increased flow provides greater delivery of oxygen and nutrients to ischemic tissues, it is often not sufficient to overcome major arterial obstruction.8,11,12
Postembryonic arteriogenesis and angiogenesis occur over a continuum of vascular adaptations (Fig. 8-1).7
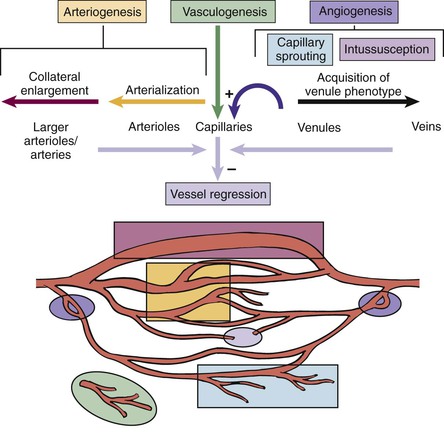
Figure 8-1 Vascular development. Postembryonic vascular remodeling occurs in response to hypoxia and fluid shear stress and involves a complex continuum of vascular adaptations. (Redrawn from Peirce SM, et al: Microvascular remodeling: a complex continuum spanning angiogenesis to arteriogenesis. Microcirculation 10:99-111, 2003, with permission from Taylor & Francis Ltd. Available at http://www.tandf.co.uk/journals.)
Arteriogenesis
Chronic, progressive arterial stenosis of an artery leads to generation of a collateral network by remodeling preexisting arterial-arteriolar connections (Fig. 8-2). Arteriogenesis is the formation of this collateral circulation, primarily in response to an increase in fluid shear stress with a contribution from increased circumferential wall stress. For example, femoral artery occlusion results in up to a 200-fold increase in shear stress in the arteriolar network.5
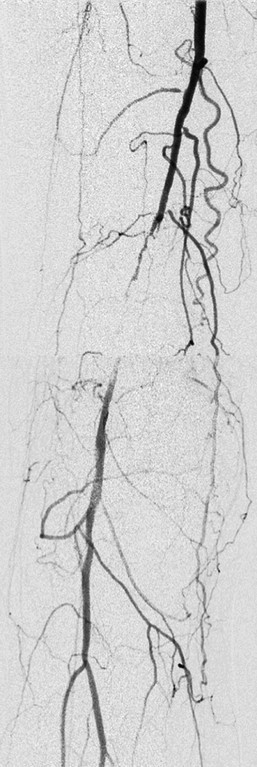
Figure 8-2 Clinical example of arteriogenesis. Digital substraction arteriography of the right lower extremity of a 72-year-old woman complaining of calf claudication. Demonstrated is a distal superficial femoral artery (SFA) occlusion with reconstitution of the distal vasculature via collaterals. Progressive stenosis of the SFA over time led to the formation of these collaterals via the process of arteriogenesis, supporting distal blood flow after the main artery occluded. These collaterals were insufficient, however, to prevent symptom development, and the patient underwent successful angioplasty and stent of the occluded vessel with good result (not shown). (Courtesy Paul DiMuzio, MD, Department of Vascular and Endovascular Surgery, Thomas Jefferson University Hospital, Philadelphia, Pennsylvania.)
Fluid shear stress is proportional to flow velocity and inversely proportional to the radius cubed, whereby small changes in radius can normalize shear stress.11 Increased fluid shear stress activates the endothelium.5,8,14 Nitric oxide (NO) is liberated by endothelial cells (via endothelial NO synthase [eNOS]) in addition to macrophages and smooth muscle cells in the adventitia (via inducible NOS). NO induces smooth muscle cell relaxation and vasodilatation beyond the arterial occlusion, thereby improving blood flow.12,13,15 NO also stimulates endothelial VEGF secretion, leading to the release of endothelial cell adhesion molecules (CAMs) and monocyte chemotactic protein-1 (MCP-1) by endothelial and smooth muscle cells.5,6,15 Both molecules mobilize to the cell surface, generating a “sticky” endothelium that enhances leukocyte attraction, adhesion, and invasion of arteriolar collaterals and periadventitia.6,8
Monocytes either attach to cell adhesion molecules to be incorporated into the lumen of the developing vessel, or accumulate in the adventitia.15 Activated monocytes release tumor necrosis factor-α (TNF-α), further enhancing monocyte attraction. Platelets also adhere, and activation stimulates growth factor and interleukin-4 production, enhancing monocyte adhesion. Monocytes stimulate production of growth factors, chemokines, and cytokines, in addition to immune cells, leading to the proliferation of collateral arterioles (Fig. 8-3).5 Macrophages contribute to remodeling of the collateral vessel by liberating proteases.6,8,9 Endothelial cell proliferation and endothelial permeability are increased, whereas vascular smooth muscle cells proliferate and change from a contractile to a proliferative phenotype.
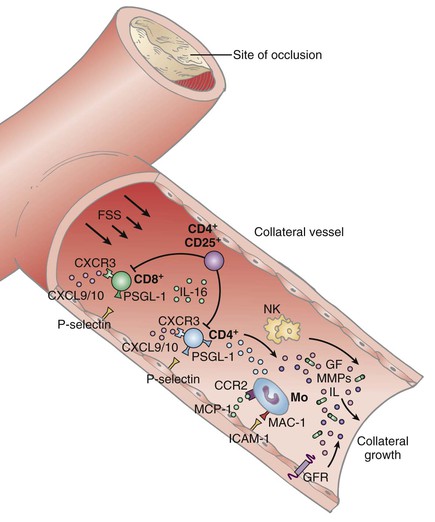
Figure 8-3 Role of immune cells in arteriogenesis. With ischemia, CD8+ T cells are attracted to the growing collateral vessel and recruit CD4+ cells, which subsequently augment arteriogenesis in association with natural killer (NK) cells. Monocytes are recruited, initiating vessel growth by synthesis of angiogenic/arteriogenic cytokines. FSS, Fluid shear stress; CXCR3, CXC chemokine receptor 3; CXCL 9/10, CXC chemokine ligand 9/10; CCR2, CC chemokine receptor 2; GF, growth factor; GFR, growth factor receptor; ICAM-1, intercellular adhesion molecule-1; MAC-1, membrane attack complex-1; MCP-1, monocyte chemotactic factor-1; MMPs, matrix metalloproteinases; Mo, monocytes; PSGL-1, P-selectin glycoprotein ligand 1. (Redrawn from Silvestre JS, et al: Post-ischaemic neovascularization and inflammation. Cardiovasc Res 78:242-249, 2008, with permission from Oxford University Press.)
Circumferential wall stress also has a role in inducing arteriogenesis. As part of a second phase of arteriogenesis, vascular smooth muscle cell growth is induced by circumferential wall stress. Increased intravascular pressure leads to smooth muscle cell proliferation and increased vessel thickness. Increased vessel thickness enables normalization of circumferential was stress at low blood pressure, which can lead to cessation of collateral vessel growth prior to the complete resolution of ischemia.11
The final phase of arteriogenesis involves “pruning” or regression of vessels. Poiseuille’s law states that flow is proportional to radius and predicts that greater flow is observed within fewer larger arterioles rather than many smaller ones. As such, the process of “pruning” leads to regression of smaller collaterals, with persistence of a few larger collateral vessels. Vessel regression is the result of proliferation of the intima that leads to occlusion of the collateral.11
Cardiovascular Risk Factors Associated with Impaired Arteriogenesis
A variety of disease states perturb arteriogenesis, leading to impairment of endothelial or macrophage function or to recruitment of endothelial progenitor cells or of growth factor production (Fig. 8-4). Aging itself affects arteriogenesis secondary to an overall decreased endothelial production of NO16 and migration by a higher rate of HIF degradation and decreased levels of VEFG, platelet-derived growth factor (PDGF), FGF-2, and chemokine signaling. Growth factor release is also impaired with aging.16,17
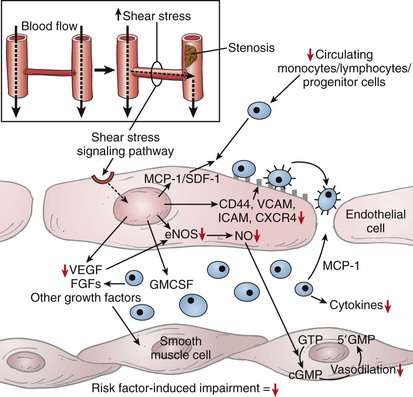
Figure 8-4 Impact of cardiovascular risk factors on arteriogenesis. With arterial stenosis or occlusion (inset), there is a drop in pressure and subsequent increase in fluid shear stress (FSS). FSS initiates activation of the intracellular signaling cascade, leading to stimulation of arteriogenesis. Red arrows indicate steps that may be negatively influenced by cardiovascular risk factors. cGMP, Cyclic guanosine monophosphate; CXCR4, CXC chemokine receptor 4; eNOS, endothelial nitric oxide synthase; FGFs, fibroplastic growth factors; GMCSF, granulocyte-macrophage colony-stimulating factor; GMP, guanosine monophosphate; GTP, guanosine triphosphate; ICAM, intercellular adhesion molecule; MCP-1, monocyte chemotactic factor-1; NO, nitric oxide; SDF, stromal cell–derived factor-1; VCAM, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor. (Redrawn from Kinnaird T, et al: Cardiovascular risk factors impair native collateral development and may impair efficacy of therapeutic interventions. Cardiovasc Res 78:257-264, 2008, with permission from Oxford University Press.)
Diabetes mellitus retards vessel formation in relation to an attenuated response to the mobilization of mononuclear cells induced by granulocyte-macrophage colony-stimulating factor (GM-CSF)18 and to impaired monocyte chemotaxis in response to VEGF.16,17,19,20 This overall decrease in circulating EPCs leads to endothelial dysfunction and poor collateral artery formation.16,20 Additionally, eNOS is inhibited by an increase in free radicals related to diabetes.17
Hypertension can have a variety of effects on arteriogenesis. Elevated blood pressure can cause an increase in fluid shear stress, which stimulates arteriogenesis. Hypertension is also associated with activation of the renin-angiotensin system and subsequent activation of arteriogenesis. Angiotensin’s role in regulating inflammatory response can lead to initiation of arteriogenesis induced by inflammation. Angiotensin also stimulates increases in circulating VEGF, platelet-derived growth factor, and FGF, all of which stimulate arteriogenesis. Conversely, activation of angiotensin by hypertension is associated with endothelial cell dysfunction as a result of increased oxidative stress due to activation of NADPH (reduced nicotinamide adenine dinucleotide phosphate) oxidase activity. The increase in oxidative stress increases reactive oxygen and superoxide levels. These oxygen radicles uncouple eNOS, thereby reducing the availability of NO.16,21,22
Hyperlipidemia affects various steps in arteriogenesis and has direct toxic effect on both endothelial cells and vascular smooth muscle cells.17 Oxidized low-density lipoprotein cholesterol (LDL) interferes with VEGF function, leading to disordered endothelial cell migration via eNOS inhibition.17,22 Additionally, endothelial cell FGF and T-lymphocyte migration is reduced, as is endothelial cell replication. Expression of FGF receptors, HIF-1, and VCAM-1 is impaired, leading to impairment of monocyte chemotaxis.23
Lastly, tobacco abuse impairs endothelial progenitor cell number, function, migration, and adherence. Monocyte migration in response to VEGF is disordered with smoking. Decreased levels of VEFG and HIF-1 further impair EPC function.16,17,22
Angiogenesis
Although arteriogenesis involves the formation of collateral arteries stimulated by fluid shear stress related to upstream arterial stenosis or occlusion, angiogenesis involves new capillary formation induced by distal tissue ischemia. Angiogenesis can occur via nonsprouting mechanisms (intussusceptive microvascular growth [IMG]) or via sprouting.24–26
Sprouting Angiogenesis
Sprouting angiogenesis involves endothelial cell projections into surrounding connective tissue. Breakdown of the basement membrane occurs, along with interendothelial junction formation, enabling endothelial cell projection.26–28 Endothelial migration occurs along the projection’s front with further sprouting, ultimately developing a complex capillary meshwork.
Sprouting angiogenesis requires specialization of cells along the migrating projection into “tip,” “stalk,” and “phalanx” cell phenotypes on the basis of the interaction of factors promoting or inhibiting angiogenesis.29–34 Tip cells are polarized migratory cells that are at the forefront of the endothelial sprout. These cells branch at the tip of the stalk as they extend filopodia toward the stimulus, yet accomplish this with hardly any proliferation. Stalk cells conversely exhibit a proliferative phenotype responsible for the lengthening of the endothelial sprout. These cells are also responsible for secretion of basement membrane along the stalk and formation of vascular lumina from the initial luminal slit.32,34,35 Additional stability to the proliferating stalk is provided by pericytes, which surround the basement membrane and provide further vessel coverage and decrease leakage from the vessel.35–37 Initially, the process of sprouting requires only minimal endothelial cell proliferation, although this demand increases with continued sprouting.29,30,35
A third cell type, the “phalanx” cells, are endothelial cells that become quiescent after completion of the vascular branch. These cells deposit basement membrane and form tight cellular junctions via increased expression of vascular endothelial cadherin (VE-cadherin). These cells are ultimately responsible for delivery of oxygen and nutrients to surrounding tissues.35,38
Regulation of Angiogenesis
Angiogenesis is initiated by ischemia, leading to increased VEGF expression. VEGF is a potent angiogenic factor, serving to encourage endothelial cell binding to VEGF receptor 2 (VEGFR-2), which promotes endothelial chemotaxis. VEGF expression induces extension of tip cells and proliferation of stalk cells with concomitant synthesis of basement membrane components.39–41 Additionally, pericytes are attracted and contribute to capillary network formation. Whereas VEGF induces sprouting, Notch signaling pathways function to limit tip migration. Notch signaling occurs by increasing expression of VEGFR-1, competitively binding VEGF and thereby limiting its availability.25,31–33,42 The balance of VEGF and Notch signaling therefore regulates sprouting-related vessel development.
Transformation to a tip cell phenotype is induced by exposure of endothelial cells to VEGF. Delta-like ligand 4 (Dll4), a Notch binding ligand, is highly expressed by tip cells, increasing sensitivity to VEGF and binding to VEGFR-2.31,32,39,43–46 Increased VEGF–VEGFR-2 binding upregulates Dll4, leading to downregulation of VEGFR-2 on adjacent endothelial cells. This process allows the tip cell to competitively maintain its position.29,33,41,47 These adjacent cells transform into a stalk cell phenotype and express Notch, which is induced by Dll-4.31,46,48 Whereas tip cells have low Notch signaling, stalk cells express higher Notch signaling and higher expression of the jagged protein-1 (JAG-1), counteracting Notch-Dll4 activity and limiting tip cell migration (Fig. 8-5).31,39,47–50
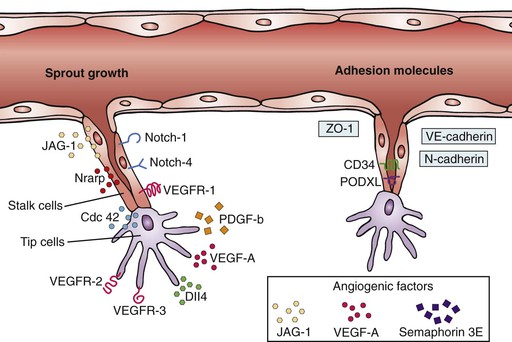
Figure 8-5 Specialization of cell phenotypes during sprouting angiogenesis. Proangiogenic and antiangiogenic factors regulate phenotypic specialization of cells into “tip,” “stalk,” and eventually quiescent “phalanx” cells. Tip cells are responsible for migration of the developing vessel and formation of filopodia, whereas the stalk cells are responsible for proliferation, lumen formation, and, ultimately, providing length to the developing vessel. Cdc 42, Cell division control protein 42 homolog; JAG-1, Jagged 1 gene; Nrarp, notch-regulated ankyrin-repeat protein; PDGF-b, platelet-derived growth factor; PODXL, podocalyxin; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; ZO-1, zonula occludens protein-1. (From Ribatti D, et al: “Sprouting angiogenesis,” a reappraisal. Dev Biol 372:157-165, 2012.)
Lumen Formation
Tubulogenesis, or lumen formation, is responsible for transforming endothelial stalks into vessels capable of carrying blood and nutrients to surrounding tissue. This process initially involves establishing endothelial cell apical-basal polarity, mediated by VE-cadherin. Apical borders face apposing cells, whereas the basal border faces the periphery. Beyond this first step, there are three proposed mechanisms to explain lumen development. The first process involves development of intracellular pinocytotic vesicles and vacuoles, which progressively fuse within the endothelial cell and then with adjacent cells, leading to lumen formation along the length of the stalk. The second mechanism is similar but involves exocytosis of these vacuoles between endothelial cells along the length of the growing stalk. These vacuoles then coalesce and form a lumen.
The third mechanism by which lumen formation may occur is by reorganization of intracellular junctions, mediated by VE-cadherin. Endothelial cells adhere to each other and become polar cells as VE-cadherin localizes CD34-sialomucins to the apical cell surface.32,51–55 The negative charges of sialomucins leads to repulsion of adjacent surfaces of the endothelial cells, inducing lumen slit formation (Fig. 8-6).32,51 As the lumen develops, the CD34-sialomucins are rearranged to the lateral surfaces of the cells and F-actin is attracted to the exposed lumen. VEGF attracts nonmuscle myosin II to the cell surface, with formation of an actinomyosin complex along the apical endothelial cell surface. This cytoskeletal interaction encourages cellular morphologic shape changes and further luminal expansion.32,51–53 Beyond tubulogenesis, further increases in lumen diameter are primarily related to fluid shear stress.29,51
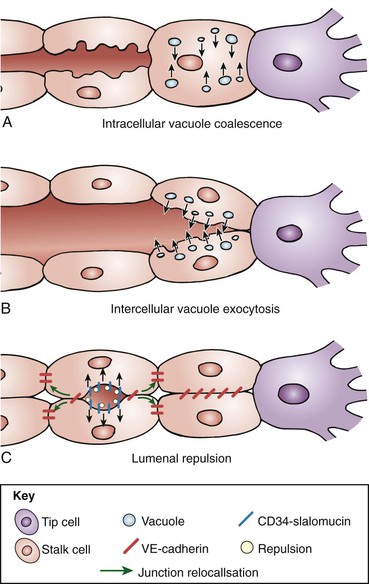
Figure 8-6 Paradigms of lumen formation in angiogenesis. A, Endothelial cells (ECs) form intracellular pinocytotic vesicles and vacuoles, which fuse within ECs and between adjacent cells, forming a lumen. B, ECs exocytose vacuoles between cells along the growing stalk. C, Polarization of ECs by VE-cadherin–mediated localization of CD34-sialomucins to the EC apical surface, leading to apical-basal polarity and subsequent repulsion and lumen formation. (From Geudens I, et al: Coordinating cell behavior during blood vessel formation. Development 138:4569-4583, 2011.)
Intussusceptive Angiogenesis
Intussusceptive angiogenesis involves the formation of transcapillary tissue pillars that fuse to form new vessels. This leads to a rapid increase in the complexity of the capillary plexus and increased surface area for gas and nutrient exchange.56–59 Initially, cell contact is created between opposing capillary cells, followed by formation of a bilayer with reorganization of endothelial cell junctions. A pillar core then develops as the result of this activity. Pericytes, myofibrils, and interstitial cells are responsible for perforating the endothelial bilayer to define discrete vessels,24,59,60 while collagen fibrils are deposited as a foundation for the developing meshwork.60 After initial capillary formation by either vasculogenesis or sprouting angiogenesis, intussusception is initiated to allow for rapid capillary expansion and remodeling by three basic mechanisms: intussusceptive microvascular growth, arborization, and intussusceptive branching remodeling.24,59–61
Intussusceptive microvascular growth initially involves formation of tissue pillars, which provides an increase in the surface area available for exchange of oxygen, carbon dioxide, and nutrients.24,58–60
The next phase, intussusceptive arborization, involves the development of “vertical pillars,” which transform well-perfused capillary segments into arterioles and venules. This process decreases the distance between arteries and veins as the capillary plexus expands. Pillars form from endothelial cell reorganization and merging of developing tissue septa (Fig. 8-7). Horizontal folds develop and lead to pillar detachment from any remaining connections, ultimately separating the feeding vessel from the capillary plexus. Hemodynamic forces therefore have a role in further development of the arterial tree.59,60,62
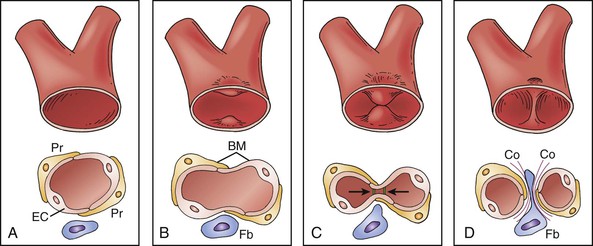
Figure 8-7 Intussusceptive growth. A and B, Impingement of endothelial cell wall into lumen, leading to creation of bilayer. This bilayer then develops perforations (C) leading to pillar formation (D). BM, Basement membrane; Co, collagen fibrils; EC, endothelial cell; Fb, fibroblasts; Pr, pericytes. (A through D from Kurz H, et al: Angiogenesis and vascular remodeling by intussusception: from form to function. News Physiol Sci 18:65-70, 2003; published by Int Union Physiol Sci/Am Physiol Soc. Entire figure is reproduced from Djonov V, Baum O, Burri PH: Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res 314:107-117, 2003. Published by Springer-Verlag.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


