Chapter 9
Arterial Aneurysms
Nathan Airhart, John A. Curci
Based on a chapter in the seventh edition by Maureen M. Tedesco and Ronald L. Dalman
Definition
An aneurysm is defined as a focal dilatation of an artery that exceeds the normal diameter by at least 50%. Aneurysms can develop at multiple locations throughout the arterial tree as a result of diverse pathologies that are often specific to that region. A “true” aneurysm results from a progressive weakening of the structural elements of the arterial wall, and both radial and longitudinal lengthening involving all three mural layers (intima, media, and adventitia). In comparison, “false” aneurysms, or “pseudoaneurysms,” form as a result of injury to the aortic wall and the flow of extraluminal blood, which is contained by surrounding tissue. Aneurysms are also classified according to their shape. “Fusiform” aneurysms are characterized by a symmetric dilatation of the complete circumference of the aortic wall. Aneurysms are classified as “saccular” when they exhibit an outpouching of only a portion of the circumference of the aortic wall.
Large elastic arteries appear to be particularly prone to aneurysmal degeneration. By far, the abdominal aortic aneurysm (AAA) is the most common type of arterial aneurysm, with an estimated prevalence of 5% to 16% in men older than 65 years. The disease in women is less common, with an estimated prevalence of 0.7% to 2.2%.1–3 AAAs can be associated with aneurysmal dilatation of the common and internal iliac arteries as well as the popliteal arteries. The AAA is the best-studied aneurysm and often serves as a reference point for studies of other, less common aneurysms.
The dissection is another arterial pathology that is often confused with primary aneurysm disease. An aortic dissection occurs acutely as a result of a mechanical separation of the layers of the aortic wall. Injury to the intima allows blood to enter the media. The laminar structure of the media is excellent for containing the normal radial force of blood pressure but poses little resistance to the intralaminar force of blood flow, resulting in predominantly longitudinal propagation of the “tear” proximally or distally. The subsequent inflammatory response to the dissection injury often leads to further structural weakening of the remaining wall, and secondary aneurysmal dilatation can then occur.
The term “atherosclerotic aneurysm” has often been used, particularly to describe aneurysms of the abdominal aorta and its branches to the lower extremities. This term is misleading in that it suggests that atherosclerosis is the cause of aneurysm disease. It is true that patients with AAA often have atherosclerosis, and population-based studies have demonstrated a strong association of coronary heart disease and peripheral atherosclerosis with AAA.3 There is very little evidence suggesting that aneurysmal degeneration is a direct consequence of atherosclerosis, however, and most research has concluded that these are independent disease processes. Supporting this hypothesis is the fact that epidemiologic data consistently demonstrate that diabetes, which is very strongly associated with atherosclerosis, has a protective effect on AAA risk. Further, there are sites along the vascular tree that are prone to atherosclerotic lesions but in which aneurysms very rarely develop (i.e., external iliac artery and superficial femoral artery).
The Aortic Wall in Health and Disease
As noted previously, the reference pathology for arterial aneurysm disease is the AAA. The normal aorta has three layers: the tunica intima, tunica media, and tunica adventitia. The tunica intima, the innermost layer of the aortic wall, is composed of endothelial cells and a thin layer of smooth muscle cells (SMCs) and connective tissue. The intima is the site of atherosclerotic disease in the aorta, and the manifestations of atherosclerosis are generally limited to this layer.
Tunica Media
The tunica media contains extracellular connective elements (elastin, collagen types I and III, proteoglycans, and glycosaminoglycans) and vascular SMCs. The architecture of the tunica media provides resistance against structural failure of the aorta. Concentric sheets of elastic membranes alternating with layers of vascular SMCs, known as lamina, are critical to the distensibility and tensile strength of the vessel. In comparison with the thoracic segment of the aorta, the abdominal aorta contains a smaller number of medial elastic lamina and a higher collagen to elastin ratio.
Elastin and Collagen
Elastin and collagen are the primary load-bearing elements of the aorta, and deficiencies or derangements of these extracellular matrix components can lead to aneurysmal disease. Normal elastic fibers are crucial to the maintenance of arterial wall integrity. Elastin, which is very distensible, functions to evenly distribute stress throughout the aortic wall. The elastic fiber is the most stable extracellular matrix component in the arterial wall, with a very long biologic half-life, typically measured in decades. During aneurysm pathogenesis, elastin fibers are dramatically depleted. This remarkable feature of the AAA media is what distinguishes the disease from other arterial pathologies and is the focus of the attempts to stabilize or restore the tensile strength of the abdominal aortic wall. Although alterations in the normal patterns of elastic fibers are observed in thoracic aortic aneurysms (TAAs), the loss of elastic fibers is not as severe as that seen in AAAs. The implication of this distinction between the two types of aortic aneurysms is not fully understood (Fig. 9-1).
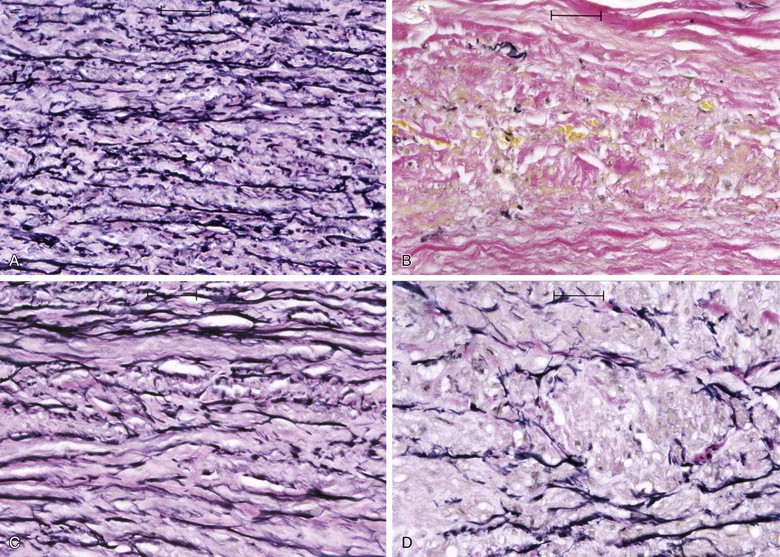
Figure 9-1 Photomicrographs (×200) of elastin-specific staining in the media of the aortic wall. They demonstrate the normal lamellar pattern of the elastin fibers in a nondilated age-matched aorta (A) and the absence of essentially any stainable elastin fibers in the abdominal aortic aneurysm (B). C and D, The media of the aneurysmal thoracic aorta, however, typically demonstrates fractures and thinning of the elastic fibers of the aortic wall.
Collagen, which has a much higher tensile strength but is less distensible than elastin, is recruited at elevated pressures, adding to resistance against further dilatation. Rupture occurs when residual and newly synthesized medial and adventitial collagen fibers fail to maintain structural integrity. The precise sequence of events leading to rupture remains unknown but almost certainly involves alterations in wall stress, medial inflammation, and proteolysis, resulting in critical reductions in mural tensile strength.
Vascular SMCs
Vascular SMCs play a very important role in normal aortic wall physiology. They are responsible for producing the components of the extracellular matrix, including elastin and collagen, and are crucial to the vascular remodeling process that occurs in response to changing stresses on the artery. The importance of normally functioning SMCs has been illustrated in studies using murine AAA models, in which repopulation of the aorta with normal vascular SMCs protected against progression of aneurysmal disease. Aneurysm pathology is associated with a depletion of medial SMCs and apparent dysfunction of those that remain. There is evidence that SMCs residing within the media of aortic aneurysms directly participate in aneurysm progression with production of matrix-degrading proteases such as matrix metalloproteinases MMP-9 and MMP-2.4,5
Adventitia
The adventitia, which is the outermost layer of the aorta, is composed of interstitial collagen fibers, fibroblasts, nerve fibers, and a network of small blood vessels that supply the adventitia and outer region of the tunica media known as the vasa vasorum. The density of the vasa vasorum decreases along the length of the aorta from aortic root to bifurcation. Speculation has existed regarding a potential relationship between reduced adventitial vasa density and the proclivity for increased aneurysm formation in the distal aorta. Evidence of aneurysm causality related to regional differences in aortic adventitial vascularity remains inconclusive, however. Increased inflammation-driven adventitial neocapillary formation, or “neovascularity,” has been recognized in surgical specimens obtained at the time of aneurysm repair (Fig. 9-2) and found to be most prominent at sites of aortic rupture.6 Though this neovascularity is clearly present, whether it actively promotes progression of aneurysm disease and rupture or simply represents evidence of progressive mural inflammation remains uncertain.
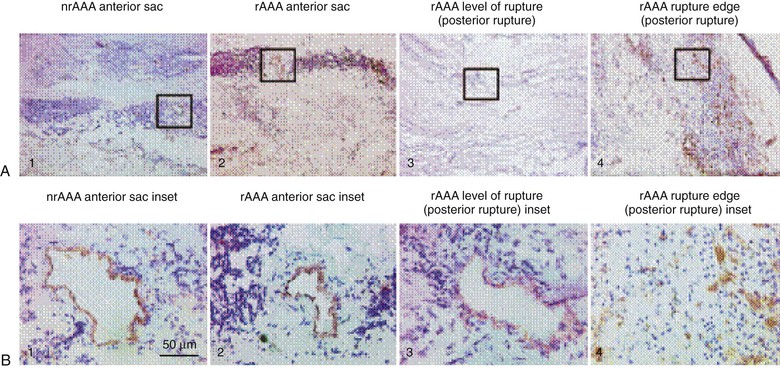
Figure 9-2 Low-power (×50) (A) and high-power (×400) (B) photomicrographs show higher-density and smaller-diameter CD31+ microvessels in the tunica media of a ruptured abdominal aortic aneurysm (rAAA) (rupture edge, posterior rupture; A4 and B4) than in a nonruptured AAA (nrAAA) anterior sac (A1 and B1), a ruptured AAA anterior sac (A2 and B2), or rAAA level of rupture (posterior rupture) (A3 and B3). (Reproduced with permission from Choke E, et al: Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol 26:2077, 2006.)
The Epidemiology of Aortic Aneurysms
Aneurysms may develop at a number of specific locations throughout the arterial tree, with corresponding risk factors, pathology, and natural history unique to each segment. Large-scale, multicenter observational series completed during the last 15 years have provided new insight into the complex and interrelated nature of aortic aneurysm pathobiology.1–3,7,8 Although epidemiologic and demographic risk factors for the development of aneurysms have been established in various populations, these relationships have proved less reliable in predicting significant clinical events such as expansion and rupture with any clinically meaningful precision. A significant reason for failing to identify predictors of disease progression is that most epidemiologic studies are based on comparisons across cohorts rather than longitudinal observations. Even longitudinal follow-up is limited by censoring of observational endpoints by surgical intervention, which obscures the natural history of advanced disease. In addition, aneurysms cannot be diagnosed until the disease process has become sufficiently advanced to result in a measurable dilatation of the artery. Pathologic studies in humans are limited to specimens obtained very late in the natural history of the disease, typically after an intervention has taken place to prevent rupture.
Risk Factors for the Development of Aortic Aneurysms
Epidemiologic risk factors for the development of abdominal aortic aneurysms include cigarette smoking, advanced age, family history, hypertension, central obesity, hypercholesterolemia, and the presence of atherosclerotic occlusive disease such as coronary artery disease.
Cigarette smoking is by far the strongest predictor of AAA in both men and women, with reported odds ratios ranging from 3.0 to 12.0.1–3 There is a strong dose-response relationship, with the risk of AAA increasing directly with the number of packs per day and number of years smoked.7 Smoking confers at least a 3.5-fold greater increase in relative risk than any other recognized AAA risk factor, and the excess prevalence associated with smoking accounts for 75% of all AAAs 4.0 cm or larger. AAA disease has the closest relationship of any disease to cigarette smoking except pulmonary malignancy and emphysema. Although cessation of smoking is associated with a decline in the risk for AAA, individuals with a remote history of smoking still have a higher risk for AAA than individuals who have never smoked. The effect of “ever smoking” is thought to be quite durable, lasting decades.9 The association of a history of “ever smoking” with the development of an aortic aneurysm in men is 2.5 times greater than the association of “ever smoking” with the development of coronary artery disease and is 3.5 times greater than the association of “ever-smoking” with cerebrovascular disease.9
Female gender, African-American race, diabetes mellitus, and regular exercise (in men) are protective against AAA disease. Although the prevalence of AAAs is much lower in women than in men, with a male-to-female ratio of approximately 4 : 1 to 6 : 1, several studies have reported that women with AAAs tend to have poorer disease outcomes than men. Abdominal aneurysms in women also exhibit an elevated risk of rupture, especially if surgical therapy is reserved for AAAs more than 5.5 cm in diameter.10 Recognized risk factors for AAA disease are summarized in Table 9-1.
Table 9-1
Epidemiologic Factors Associated with Abdominal Aortic Aneurysms
| Factor | Odds Ratio |
| Hyperhomocysteinemia | 7.8 |
| Current smoker | 7.4 |
| Ever smoked | 5.1 |
| Hernia | 3.9 |
| Low vitamin B6 levels | 3.75 |
| Age 75-84 years | 3.3 |
| Family history of abdominal aortic aneurysms | 1.9 |
| Age (per 7-year increase) | 1.7 |
| Symptomatic atherosclerosis | 1.7 |
| Hypercholesterolemia | 1.4 |
| Serum resistin concentration | 1.53 |
| Waist-to-hip ratio | 1.22 |
| Waist circumference | 1.14 |
| Black race | 0.5 |
| Diabetes mellitus | 0.5 |
| Female sex | 0.2 |
The negative association observed between diabetes mellitus and AAA has been a focus of much attention. A number of studies have demonstrated this apparent protective effect, with a decreased incidence of AAA disease in diabetics as well as decreased aneurysm growth and rupture rates. These observations reinforce the idea that the pathogenesis of AAA is independent from the development of atherosclerosis and aorto-occlusive disease.11
Risk for Expansion and Rupture of Aortic Aneurysms
Once aneurysm pathology is initiated, genetic and environmental factors influence further dilatation of the aorta and progression toward rupture. Although rates of enlargement vary with time and aortic diameter, the average abdominal aortic aneurysm grows at the rate of 2 to 3 mm/yr. Smoking is a major risk factor for progression and rupture of AAA. A meta-analysis using data from patients with small AAAs demonstrated that the rate of expansion increases with current smoking status by approximately 20%. This meta-analysis reported that current smoking status doubled the risk of rupture in individuals with AAAs (hazard ratio 2.02; 95% confidence interval [CI] 1.33-3.06).12
The baseline diameter of an aneurysm strongly influences its growth rate (larger AAAs grow more rapidly). Data from the UK small aneurysm trial demonstrated that AAAs 5 cm in diameter grow approximately 70% faster than those 4 cm in diameter.13 As the size of the aneurysm increases, so does the risk of rupture. Several large population studies have demonstrated that the risk of aneurysm rupture is very low (0%-2.5% after 5 years) for aneurysms less than 5.0 cm in diameter. For aneurysms measuring more than 5.0 cm, the 5-year risk of rupture rises well above 20%.14,15 Data from the UK Small Aneurysm Trial and the Aneurysm Detection and Management (ADAM) study did not show an advantage of surgical repair for AAA measuring less than 5.5 cm.2,13 For this reason, elective repair of AAAs is most often indicated for aneurysms larger than 5.5 cm.
After initial diagnosis, risk for expansion and rupture of AAAs differs between men and women. Multiple studies have demonstrated that women have a higher risk of AAA rupture than men.1,16 Mortality following repair of AAA is also higher in women than in men, a difference that is most pronounced with endovascular repair (EVAR).17 There are several proposed causes of this discrepancy. The first is that women often have smaller normal aortic diameters than men, so the absolute indication for surgical repair of an AAA (5.5 cm) may reflect a later stage of disease development in women. Another factor relates to the reduced cardiovascular disease awareness in women compared with men, which may lead to delayed diagnosis and intervention. Finally, inferior outcomes following endovascular repair of AAAs in women may relate to the actual endovascular devices used to stabilize the aorta, because these devices are designed for and tested based mainly in male anatomy.17
Recognized AAA risk factors, including peripheral vascular disease (as indicated by reduced ankle-brachial indices), hypertension, and hyperlipidemia, have not been consistently associated with rates of expansion. As mentioned, the presence of diabetes mellitus has a protective effect on aneurysm growth (reduction by 30%).2,3,11–13
Pathophysiology of the Aneurysm Wall
Because abdominal aortic aneurysms are the most common type of aneurysm disease, research efforts to determine the pathobiology of arterial aneurysms have traditionally focused on AAAs. The pathobiology of AAAs also serves as a useful reference point for studies on other types of aneurysms. Since the first investigations on AAAs as a disease process potentially distinct from atherosclerosis more than 20 years ago, there has been an exponential growth in our understanding of the pathobiology underlying AAAs. This growth has been due to increasingly detailed study of human AAA tissues and has been facilitated by development of successful animal models.
Animal Models of Aneurysm Pathogenesis
The aneurysm is, by nature, a degenerative disease that is evident only when the structural failure leads to dilatation. Therefore, the advanced state of aneurysmal degeneration present in most surgical specimens provides little opportunity for analytic insight into the mechanisms responsible for initiation or early progression of disease. Toward the goal of understanding the mechanisms that result in the late-stage disease seen in human pathology, a variety of experimental aneurysms have been created in rodent models that share important pathologic, cellular, and humoral features with human AAA disease.19,20
Temporary introduction of porcine pancreatic elastase into the infrarenal aorta reliably promotes aneurysmal degeneration in mice, rats, rabbits, and pigs. After a very brief dwell time within an isolated segment of murine aorta, porcine pancreatic elastase alone results in limited damage to the aorta. However, progressive medial elastolysis, transmural mononuclear infiltration, and SMC apoptosis ensues over days to weeks (Fig. 9-3). The most important feature of this model is the transient nature of the elastase effect; within 24 hours no trace of exogenous elastase is present, whereas peak inflammation occurs days later. Relative strengths of this model include the development of circumferential aneurysmal degeneration in vessels of varying size and precise localization in the infrarenal segment of the aorta. Although dilatation is very consistently achievable, the aneurysms in these models nearly always stabilize structurally and do not proceed to rupture. Other limitations include the need for direct manipulation and injury to the aorta.
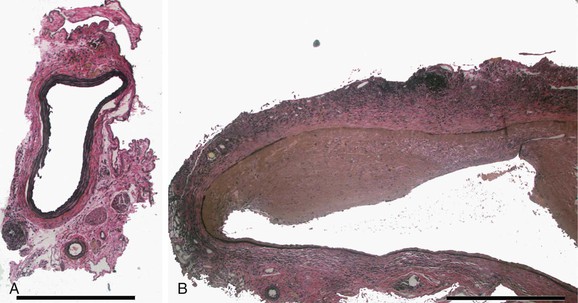
Figure 9-3 Histologic changes after elastase perfusion. Photomicrographs at the same magnification of a normal preperfusion mouse aorta (A) and a mouse aorta perfused with active porcine pancreatic elastase 14 days after perfusion (B). After elastase perfusion, there is a fusiform aneurysm of the aorta with extensive inflammation and medial elastin loss.
Long-term subcutaneous infusion of angiotensin II (Ang II) in congenitally hyperlipidemic mice deficient in either apolipoprotein E or low-density lipoprotein receptors promotes AAA development in the perivisceral segment of the mouse aorta. The dilatation occurs over the course of several days with only mild increases in blood pressure; aneurysm formation in these mice is frequently preceded by focal aortic dissections stimulating inflammation, degradation of elastin, and new vessel formation (Fig. 9-4).20 Relative strengths of this model include the co-occurrence of fatty streaks and early atherosclerosis in the proximal aorta and distal arterial beds (as is commonly present in the human condition), as well as the development in the absence of direct aortic manipulation. Relative limitations include the uncertain relevance of hyperlipidemia to human AAA disease, the typically suprarenal location of the resulting aneurysms (with uncertainty regarding the “appropriate” location for AAAs in quadruped mammalian models), and the failure of the end-stage diseased pathology to closely resemble the appearance of human pathology.
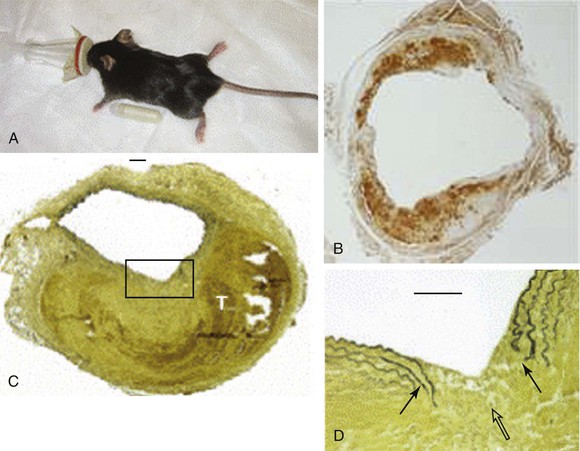
Figure 9-4 Angiotensin II model of abdominal aortic aneurysm disease. A, An osmotic pump filled with angiotensin II is inserted into the subcutaneous tissue of the mouse for continuous delivery. B, Mac-2 stain of an angiotensin II–induced aneurysm illustrating transmural inflammation. C, Verhoeff-van Giessen–stained section demonstrating elastin rupture; D, enlarged view of square in C. The medial elastin bands (black arrows in D) display an area of complete rupture (open arrow in D) leading to thrombus formation (T in C). (Reprinted with permission from Gavrila D, et al: Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 25:1671, 2005.)
The calcium chloride model induces AAA formation through periaortic (in rats and mice) or intraaortic (in rabbits) application of 250 mmol/L of CaCl2. In this model, focal aortic enlargement is induced through a chemical injury initiated from outside the adventitia. Though this method is reliable and focally reproducible, the AAAs it creates tend to stabilize after several days and rarely proceed to rupture.
Each of the three models incorporates features that are remarkably similar to individual facets of clinical disease, but no one model faithfully recreates the human condition in its entirety. Other, less frequently used models, such as the xenograft transplant model, help to further elucidate the mechanisms of aneurysmal degeneration. Understanding of the specific features and limitations inherent in each model is essential for interpreting the significance and potential generalizability of results obtained from any one model alone. Familiarity with the limitations inherent in rodent modeling is also critical for understanding why so many promising therapeutic strategies found to limit progression of experimental aneurysms have failed to make the transition to effective clinical treatments. Readers interested in a more in-depth comparison between available AAA model systems are referred to a comprehensive review by Daugherty and Cassis.19
Proteolytic Activity within the Aneurysm Wall
The medial destruction characteristic of the abdominal aortic aneurysm is remarkable for the near-complete elimination of the normal structural elements, particularly the typical elastic fiber sheets. Because the elastin is normally incredibly durable, the investigations into the pathophysiology of the AAA have focused on the limited number of enzymatic processes capable of elastolysis. Elastolytic enzymes noted to be substantially elevated within the aneurysm wall include neutrophil elastase as well as several members of the MMP class. Subsequently, a number of other elastolytic enzymes have also been identified in aneurysm tissue and implicated in the destructive process. However, the biology of MMPs in the AAA have been very well characterized and likely play a supporting, if not central, role in the disease.
Matrix Metalloproteinases and AAA
MMPs are a family of extracellular matrix–degrading enzymes essential for a range of homeostatic physiologic processes, including wound healing, angiogenesis, tissue remodeling, and bone resorption. The active sites of MMPs have substantially similar amino acid sequences, contain a zinc ion vital for their enzymatic activity, and are inhibited by chelating agents. In vivo, they are inhibited by the expression and local release of biologic inhibitors of MMP activity such as tissue inhibitors of metalloproteinases (TIMPs). Pro-MMPs are secreted by neutrophils, macrophages, fibroblasts, and vascular SMCs. Enzymatic cleavage and activation are catalyzed by extracellular proteases such as plasmin, plasminogen activators, and other MMPs. MMPs are controlled at several levels, including the induction and suppression of MMP gene transcription, extracellular activation, and interaction with natural inhibitors. They were originally categorized according to substrate (collagenase, elastase, and other constituents), but later classifications are numbered in recognition of the considerable substrate overlap that exists between functionally distinct MMPs.
Several MMPs have been implicated in AAA disease, including types 1, 2, 3, 8, 9, 12, and 13 and membrane type 1 MMP (MT1-MMP), with molecular weights ranging from 52 to 92 kD (Table 9-2).21–35 Targeted deletions of genes encoding for TIMP expression lead to the development of larger aneurysms, thus suggesting the possibility that aneurysmal disease may also result from a local or systemic imbalance between TIMP and MMP production. The relationship between MMP and TIMP production is almost certainly more nuanced in the human disease condition; tissue analysis has consistently demonstrated increased TIMP production in AAA surgical specimens.
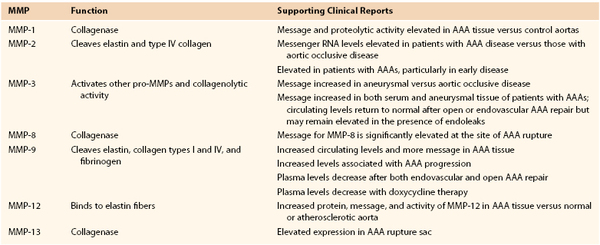
MMP-9, also known as gelatinase B or 92-kD gelatinase, cleaves elastin, collagen types I and IV, and fibrinogen. Several in vitro studies have demonstrated that MMP-9 is the predominant gelatinase produced in AAA tissue. It is more highly expressed in aneurysmal compared to normal aortic tissue, with infiltrating macrophages being its dominant source. However, it has also been shown that vascular SMCs within the tunica media, as well as adventitial fibroblasts, are capable of expressing MMP-9, especially under inflammatory conditions. Plasma MMP-9 levels are also increased in AAA disease. Patients with intermediate-sized AAAs (between 5 and 6.9 cm) have higher circulating levels than patients with smaller (<4 cm) or larger (>7 cm) AAAs.29 MMP-9 knockout mice are protected against aneurysm formation and exhibit morphologic preservation of elastic lamella.30
Like MMP-9, MMP-2 is capable of degrading elastin and type IV collagen. It is constitutively expressed by SMCs and fibroblasts. Aortic samples obtained from smaller aneurysms demonstrate relatively more MMP-2 than MMP-9 activity, suggesting that increased MMP-2 activity may represent an early event in the time course of AAA pathogenesis. Like mice deficient in MMP-9 production, those deficient in MMP-2 production are protected from aneurysm formation. Experimental data also suggest that both MMP-2 and MMP-9 need to be present and active for maximal aneurysm progression to be achieved, thus implying a synergistic and codependent relationship.31
One of the most potent elastolytic enzymes in this class is MMP-12, or human macrophage elastase (HME). The murine analog was shown to be critical to smoke-induced emphysema in mice. In an evaluation of human aortic aneurysm specimens, the enzyme and its activity were found to be substantially increased. Moreover, the enzyme was specifically localized by immunohistology to the residual elastic fibers within the wall of the aneurysm (Fig. 9-5).32
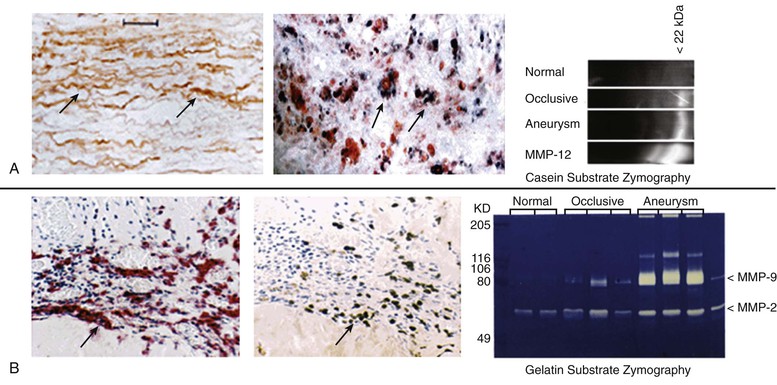
Figure 9-5 Histologic and zymographic evidence of representative active elastolytic enzymes of the matrix metalloprotease (MMP) family in human aneurysm wall. A, The enzyme MMP-12 is localized by immunohistology to the residual elastic fibers in the wall of the abdominal aortic aneurysm by in-situ hybridization to the inflammatory cells of the aortic wall, and by casein zymography. B, The presence of MMP-9 is also seen in the wall of the aorta by immunohistology, in-situ hybridization, and gelatin zymography.
Other Proteinases in Abdominal Aortic Aneurysms
Several proteases from different classes have been demonstrated to contribute to AAA disease either through direct destruction of extracellular matrix or by activating latent elastolytic MMPs. Cathepsins are cysteine proteases that catalyze elastin degradation and depletion in experimental AAA disease. Increased amounts of cathepsins K, L, and S are present in the human aortic wall. As is the case for several key MMPs, human aortic aneurysm specimens show a greater activity of cathepsins B, H, L, and S than do surgical specimens obtained from patients with occlusive aortic disease. Expression of cystatin C, a cysteine protease inhibitor, is reduced in human and experimental aneurysm tissue. Targeted deletion of cystatin C in experimental aneurysm disease increases aneurysm size and the rate of expansion.
The cysteine protease dipeptidyl peptidase I (DPPI) appears to facilitate model aneurysm development through the recruitment of neutrophils, which appear to be critical for the development of aneurysms in the elastase model.36 Expression of tenascin-X, an extracellular matrix protein absent in Ehlers-Danlos syndrome, is also markedly diminished in AAA tissue despite elevated serum levels. Serine proteases, particularly those from the plasminogen activator family, urokinase plasminogen activator (u-PA) and tissue plasminogen activator (t-PA), have also been demonstrated to be involved in AAA pathogenesis. These enzymes cleave plasminogen, forming plasmin, which is capable of directly degrading extracellular matrix components and also of activating metalloproteinases.37
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


