Arrhythmias
Cardiac arrhythmias were first noted in antiquity, when physicians began to palpate the arterial pulse. Hippocrates noted that a slow pulse in elderly men heralded sudden death, and Galen predicted the impending death of an apparently healthy physician who had observed that his pulse was irregular. These and similar observations were systematized in the late 19th century, when smoked drum kymograph recordings of arterial and venous pulsations were used to define simple arrhythmias. Knowledge of clinical arrhythmias advanced rapidly after Einthoven’s invention of the string galvanometer at the beginning of the 20th century, which made it possible to record abnormalities of cardiac rate and rhythm in experimental animals and humans. By the middle of the 20th century, electrocardiographic analyses characterized many of the pathophysiological mechanisms that operate in patients with arrhythmias (Katz and Pick, 1956). Intracardiac recordings, which became available in the 1960s, along with microelectrode recordings of intracellular potentials and patch clamp measurements of the opening and closing of single ion channels, greatly advanced our understanding of arrhythmogenic mechanisms. Molecular biology, which is providing the molecular structures of the voltage-gated ion channels and the cellular components that regulate their behavior (see Chapters 13 to 15), is now adding a new dimension to our understanding of arrhythmogenic mechanisms.
A Simple Classification of Arrhythmias
Normal heart rate is defined as 60 to 100 beats/min (for simplicity, only the rates, e.g., “60 to 100,” are stated in the remainder of this text); this range was chosen because these limits correspond to five and three large (0.2 s) “boxes” in the standard ECG. Arrhythmias, which can occur when the heart beats too slowly (bradycardias), too rapidly (tachycardias), and/or irregularly (Table 16-1), are generally classified on the basis of the structure where the arrhythmia is believed to have originated and the mechanism that is presumed to have disturbed the rhythm (Table 16-2). Tachycardias, which by definition occur when heart rate is sustained at rates greater than 100, are generally subdivided into supraventricular and ventricular tachycardias. The former arise above the bifurcation of the AV bundle, and so are conducted normally through the two bundle branches into the ventricles, while ventricular tachycardias distort and prolong the QRS because the ventricles are not depolarized simultaneously.
It might seem logical that the causes of tachycardias differ fundamentally from the causes of bradycardias, and that mechanisms that cause the heart to beat too rapidly are opposite those that slow the heart. However, many mechanisms that are responsible for bradyarrhythmias also cause tachyarrhythmias, so that as is often true, what seems obvious is wrong!
Table 16-1 A Simple Classification of Clinical Arrhythmias | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Mechanisms Responsible for Bradyarrhythmias
The usual causes of a slow pulse are reduced pacemaker activity (chronotropy) and depressed conduction (dromotropy). The former is caused by changes in the ionic currents responsible for SA node depolarization (see Chapter 14), while the latter, generally called block, results from impaired impulse conduction from the SA node to the ventricles.
Slowed Pacemaker Activity
The most common cause of slowing of the SA node pacemaker, called sinus bradycardia, is excessive parasympathetic (vagal) tone. Although a heart rate below 60 is defined as abnormal, sinus bradycardia is often seen in normal individuals, especially when training has increased
vagal tone. Non-cardiac disease, like hypothyroidism, can also slow the sinus pacemaker, as can drugs like β-adrenergic blockers and some L-type calcium channel blockers. Sinus bradycardia can also be caused by a condition, commonly seen in the elderly, that has the sibilant name sick sinus syndrome; because this syndrome can be accompanied by supraventricular tachycardias, it is sometimes called the bradycardia-tachycardia syndrome. When severe, sinus slowing can cause syncope, but rarely sudden death.
vagal tone. Non-cardiac disease, like hypothyroidism, can also slow the sinus pacemaker, as can drugs like β-adrenergic blockers and some L-type calcium channel blockers. Sinus bradycardia can also be caused by a condition, commonly seen in the elderly, that has the sibilant name sick sinus syndrome; because this syndrome can be accompanied by supraventricular tachycardias, it is sometimes called the bradycardia-tachycardia syndrome. When severe, sinus slowing can cause syncope, but rarely sudden death.
Table 16-2 A Simple Classification of Arrhythmogenic Mechanisms | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Abnormal slowing of “lower” pacemakers in the AV node and His-Purkinje system (AV bundle, bundle branches, fascicles) cannot cause a bradycardia as long as the ventricles are depolarized by impulses conducted from a normally functioning SA node. Failure of lower pacemakers is therefore apparent only if sinus rate is markedly slowed or conduction is blocked.
Table 16-3 Sites of Conduction Block | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Impaired Conduction (Block)
Impaired impulse conduction from the SA node to the ventricles causes ventricular beats to be absent (“dropped”). This abnormality, called block, can occur in the SA and AV nodes, where small action potentials and a low safety factor makes conduction physiologically precarious (Chapter 14), and in the AV bundle and bundle branches (Table 16-3). In the latter, in spite of the large action potentials, conduction is anatomically precarious because these conducting structures can be damaged or destroyed by disease.
Slowing of Impulse Conduction
Conduction can be slowed by decreased action potential amplitude, reduced rate of depolarization, a higher threshold, and increased internal resistance (Table 16-4). Action potential amplitude and rate of depolarization are proportional to the open probability and number of active plasma membrane channels that carry the inward currents responsible for the action
potential upstroke. Threshold reflects the ability of an approaching wave of depolarization to open these channels, and internal resistance is determined largely by the number of open gap junction channels in the intercalated discs. Changes in membrane capacitance, electrical resistance across the membrane bilayer, and the conductivity of the extracellular space also influence conduction velocity, but are of minor importance in modifying conduction velocity in the heart.
potential upstroke. Threshold reflects the ability of an approaching wave of depolarization to open these channels, and internal resistance is determined largely by the number of open gap junction channels in the intercalated discs. Changes in membrane capacitance, electrical resistance across the membrane bilayer, and the conductivity of the extracellular space also influence conduction velocity, but are of minor importance in modifying conduction velocity in the heart.
Table 16-4 Major Determinants of Conduction Velocity in the Heart | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Conduction is accelerated by increasing the rate of opening of depolarizing channels because the more rapid development of inward current shortens the time needed for an approaching action potential to bring resting tissue to threshold. Similarly, increased action potential amplitude is able to depolarize resting cells further ahead of an approaching wave of depolarization (Fig. 16-1A). Opening of gap junction channels in the intercalated disc, which decreases internal resistance, speeds conduction by facilitating the propagation of the currents generated by an approaching wave of depolarization (Fig. 16-1B), while a lower threshold increases the ability of an approaching wave of depolarization to initiate a propagated action potential (Fig. 16-1C).
The four determinants of conduction velocity listed in Table 16-4 can be likened to a row of falling dominoes (Fig. 16-2), where increasing the height of the dominoes, like increasing action potential amplitude, accelerates “impulse” transmission by allowing each falling domino to topple dominoes further ahead in the row. Increasing the velocity at which each domino falls (as would occur if the dominoes were moved to the surface of Jupiter), like increasing the rate of depolarization, accelerates transmission. Placing the row of dominoes in a vacuum, like reducing longitudinal resistance, increases conduction velocity by accelerating their fall, and if each domino was tipped slightly to reduce the time needed for it to fall, the increased “tippiness,” like a decrease in threshold, would increase the velocity of propagation.
These principles explain the different conduction velocities in various regions of the heart. Rapid conduction in the working cells of the atria and ventricles and the cells in His-Purkinje system reflects their high content of sodium channels that open rapidly and carry large depolarizing currents, and their large diameter and high number of connexin channels that reduce internal resistance. Conversely, the slower conduction in the SA and AV nodes is due to the slower opening, lower conductance, and smaller number of the calcium channels that depolarize these cells, along with a high internal resistance caused by their small size and few gap junctions. In diseased hearts, changes in many of these variables play a major role in producing the arrhythmias that often kill patients with heart disease (see Chapters 17 and 18).
Decremental Conduction and Block
Decremental conduction refers to a progressive decrease in conduction velocity when an impulse enters a region of slow conduction (Fig. 16-3). This can occur if conduction is slowed by a decrease in action potential amplitude and rate of rise, as shown in Figure 16-3, and by an increase in threshold or internal resistance. If the wave of depolarization is able to pass through the area of slow conduction, it can emerge as a normally sized action potential (Fig. 16-3A) so that conduction will be delayed but not completely blocked. However, conduction will be interrupted if the ability to generate a propagated action potential is so severely depressed that the
action potential cannot serve as an effective stimulus to the excitable tissue ahead (Fig. 16-3B). Decremental conduction is not always abnormal. For example, decremental conduction is a normal property of the AV node where cells have a small diameter, few gap junctions, and slowly rising calcium-dependent action potentials.
action potential cannot serve as an effective stimulus to the excitable tissue ahead (Fig. 16-3B). Decremental conduction is not always abnormal. For example, decremental conduction is a normal property of the AV node where cells have a small diameter, few gap junctions, and slowly rising calcium-dependent action potentials.
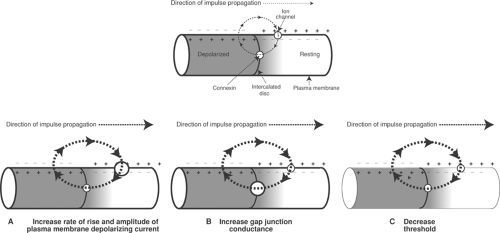 Fig. 16-1: Determinants of conduction velocity in cardiac muscle. Conduction is from left to right; depolarized tissue is shaded and resting tissue unshaded. Conduction velocity is accelerated when the magnitude and rate of rise of the inward currents carried by plasma membrane ion channels is increased (A), when internal resistance is reduced by opening of connexin channels in the intercalated discs (B), and when threshold is reduced (shown as a thinner plasma membrane in C). |
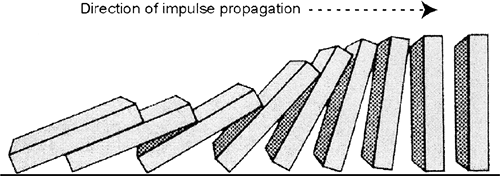 Fig. 16-2: Propagation of an action potential viewed as a row of dominoes falling from left to right. Conduction is accelerated when the height of the dominoes is increased and/or each domino falls more rapidly, when less resistance is encountered by the falling dominoes, and when less inertia is needed to tip each domino. |
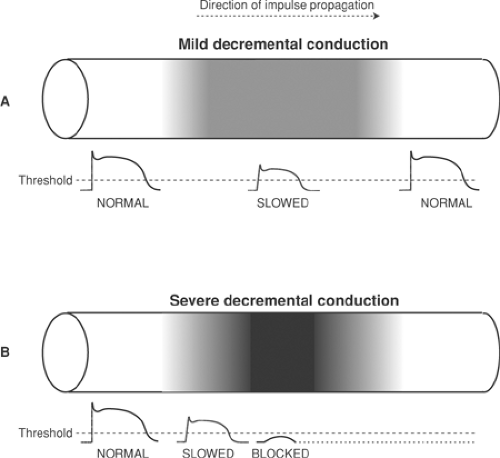 Fig. 16-3: Decremental conduction caused when an action potential enters a region where the amplitude and rate of rise of the action potential are decreased (shaded). A: Conduction of an action potential is slowed when it becomes smaller and more slowly rising in a region of decremental conduction. After the impulse has crossed the region of decremental conduction and has emerged in a region where the amplitude and rate of rise become normal, conduction velocity returns to normal. B: When an impulse enters a region where the action potential no longer reaches threshold, and so cannot initiate a propagated action potential, conduction is blocked. |
Unidirectional Block
The term unidirectional block means what it implies: block of conduction, but in only one direction. Although this behavior might seem intuitively to be unusual, it is not. Unidirectional block is common, and it is a normal property of the AV node where antegrade (forward) conduction from atria to ventricles is usually more rapid than retrograde (backward) conduction from ventricles to atria. Because unidirectional block can disorganize impulse propagation and cause conduction to fail, it plays a major role in the genesis of arrhythmias (see below).
The classical model for unidirectional block is a strand of myocardial tissue that is compressed by a wedge-shaped wooden block, which causes an asymmetrical impairment of conduction. Depending on how much pressure is applied to the wedge, the block can be unidirectional (Fig. 16-4B) or complete (Fig. 16-4C). The mechanism responsible for unidirectional block is shown in Figure 16-5, where the ability of different regions of a 10 cm long strand of myocardium to initiate a regenerative response is plotted on the ordinate. Normal action potentials in the uncompressed regions, from 0 to 2 cm and from 8 to 10 cm, can activate resting tissue up to 2 cm ahead, while between 2 and 3 cm, under the point of the wedge, the cells are so severely damaged as to have completely lost the ability to initiate a propagated action potential. Action potentials in the less depressed region, between 3 and 8 cm, can activate resting tissue but, for only short distances ahead, depending on the degree of compression. The antegrade direction in this figure is defined as from left to right, and the retrograde direction from right to left.
An action potential initiated by a stimulus on the left side, at 0 cm, is conducted in the antegrade direction (Fig. 16-5B). Normal impulses in the uncompressed tissue, between 0 and 2 cm, generate a wave of depolarization that can cross the severely depressed area between 2 and 3 cm.
Even though propagated impulses cannot be generated in the severely depressed area, electrotonic spread of current from the normal region can generate a regenerative response 2 cm ahead (arrow a), which initiates an action potential at 4 cm. Although the tissue at 4 cm is depressed, it is able to depolarize tissue a short distance (0.4 cm) ahead, at 4.4 cm (arrow b); here the action potentials are less depressed and so conduct 0.6 cm to the right, reaching the tissue at 5.0 cm (arrow c). At 5.0 cm the ability to conduct is less depressed, so that the impulse continues to propagate to the right, gaining speed as it emerges from the depressed area until
it reaches uncompressed tissue at 8 cm. Because conduction velocity is directly proportional to the lengths of the arrows in Figure 16-5B, the antegrade impulse reaches normal tissue to the right after a delay.
Even though propagated impulses cannot be generated in the severely depressed area, electrotonic spread of current from the normal region can generate a regenerative response 2 cm ahead (arrow a), which initiates an action potential at 4 cm. Although the tissue at 4 cm is depressed, it is able to depolarize tissue a short distance (0.4 cm) ahead, at 4.4 cm (arrow b); here the action potentials are less depressed and so conduct 0.6 cm to the right, reaching the tissue at 5.0 cm (arrow c). At 5.0 cm the ability to conduct is less depressed, so that the impulse continues to propagate to the right, gaining speed as it emerges from the depressed area until
it reaches uncompressed tissue at 8 cm. Because conduction velocity is directly proportional to the lengths of the arrows in Figure 16-5B, the antegrade impulse reaches normal tissue to the right after a delay.
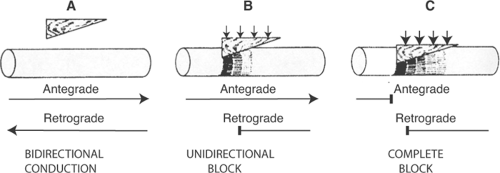 Fig. 16-4: Unidirectional block produced by compression of a strand of cardiac muscle by a wedge-shaped wooden block (darker shading indicates a greater degree of injury). A: Bidirectional conduction in the normal, uncompressed tissue. B: Moderate compression causes unidirectional block in which conduction can proceed from left to right (defined here as the antegrade direction) but not from right to left (retrograde block). C: Severe compression blocks conduction in both directions. |
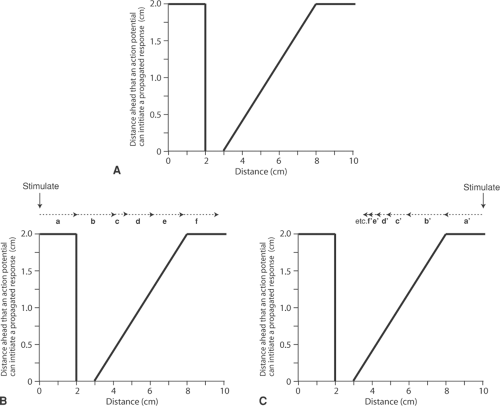 Fig. 16-5: Unidirectional block. A: Distribution of the ability of the moderately compressed strand of cardiac muscle shown in Figure 16-4B to generate a propagated action potential. Normal tissue (at 0 to 2 and 8 to 10 cm) can activate resting muscle up to 2 cm ahead. The ability of the tissue at 2 to 3 cm to initiate a propagated action potential is completely lost, and is depressed from 3 to 8 cm. B: Antegrade conduction. An impulse entering from the left is able to cross the most severely depressed region, where the ability to initiate a propagated action potential has been lost, although antegrade conduction (left to right) is delayed. C: Retrograde block. An impulse entering from the right encounters increasingly impaired conduction, so that the impulse becomes progressively less able to initiate an action potential. As the impulse approaches the most severely depressed tissue (from 2 to 3 cm), the responses become so small that they cannot excite the resting tissue ahead; as a result, retrograde conduction (right to left) is blocked. The dotted arrows in B and C depict the distance ahead that action potentials generated at the tail of the arrow can be propagated. |
When an action potential is initiated by stimulating the tissue on the right side, at 10 cm, rather than at 0 cm, retrograde conduction is quite different (Fig. 16-5C). At 8 cm, although the impulse encounters damaged tissue and conduction begins to slow, it can still be conducted to the left, and so depolarizes the tissue 2 cm ahead, at 6 cm, where action potentials can propagate only 1.2 cm (arrow a’). The next area to be depolarized is therefore at 4.8 cm (the head of arrow b’), where the action potential can conduct only 0.8 cm to reach the tissue at 4 cm (arrow c’). Here, the action potential conducts an even shorter distance (arrow d’). Propagation continues to slow, as evidenced by the shorter arrows, until the impulse reaches the severely depressed area at 3 cm where, because propagated action potentials can no longer be generated, retrograde conduction ceases.
Areas of unidirectional block, such as that shown in Figure 16-5, are common in diseased hearts, where non-uniform depression of conduction velocity can be caused by asymmetrical decreases in action potential amplitude, rate of depolarization, number of open gap junction channels, and excitability.
Tachyarrhythmias
Extra beats and tachycardias are generally described in terms of their clinical manifestations (Table 16-1). A single early beat is a premature systole and a series of premature systoles is a tachycardia. Very rapid regular depolarization of the atria or ventricles is called flutter, while rapid irregular depolarization, where there is no effective mechanical response, is fibrillation. Tachyarrhythmias can result from accelerated pacemaker activity, reentry, and triggered depolarizations (Table 16-2).
Accelerated Pacemaker Activity
Accelerated firing of pacemaker cells in the SA node causes sinus tachycardia, which is often a normal response to increased sympathetic tone or other extracardiac factors. Accelerated pacemaker discharge in “lower” pacemakers causes junctional tachycardias and accelerated idioventricular rhythms.
Reentry
Reentrant arrhythmias, which occur when a single impulse gives rise to two or more propagated responses, can be caused by both pathophysiological and anatomical mechanisms (Table 16-2). The former include conduction abnormalities, and heterogeneities in resting potential, action potential amplitude, and refractoriness. Anatomical causes of reentry can result from asymmetric tissue damage and abnormal conducting structures.
Slow Conduction (Decremental Conduction and Unidirectional Block)
The mechanism by which slow conduction causes a reentrant premature systole or tachycardia can be understood by examining the junction where an impulse conducted by a Purkinje fiber activates the ventricular myocardium (Fig. 16-6). When a wave of depolarization (labeled 1 in Fig. 16-6) reaches the proximal end of an area where unidirectional block prevents antegrade
conduction (shaded area in Fig. 16-6), the impulse cannot cross the depressed region. However, if propagation through normal regions of the heart brings this impulse to the distal end of the area of unidirectional block after a delay that is long enough to allow the depressed tissue to recover its excitability, the wave of depolarization can be conducted through the depressed area in the retrograde direction. This can allow the impulse to depolarize (reenter) the proximal region and generate a second propagated wave of depolarization (labeled 2 in Fig. 16-6). Conduction of the retrograde impulse often increases the refractoriness of the depressed area so as to block conduction in both directions, which terminates the reentry. However, repeated conduction through a reentrant circuit can generate a sustained tachycardia (see below).
conduction (shaded area in Fig. 16-6), the impulse cannot cross the depressed region. However, if propagation through normal regions of the heart brings this impulse to the distal end of the area of unidirectional block after a delay that is long enough to allow the depressed tissue to recover its excitability, the wave of depolarization can be conducted through the depressed area in the retrograde direction. This can allow the impulse to depolarize (reenter) the proximal region and generate a second propagated wave of depolarization (labeled 2 in Fig. 16-6). Conduction of the retrograde impulse often increases the refractoriness of the depressed area so as to block conduction in both directions, which terminates the reentry. However, repeated conduction through a reentrant circuit can generate a sustained tachycardia (see below).
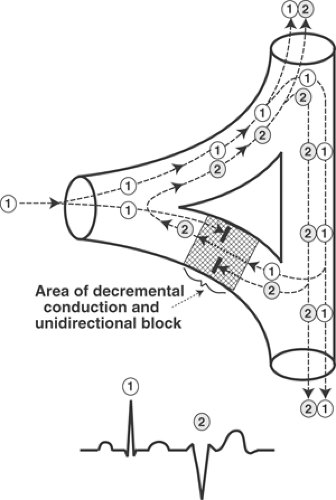 Fig. 16-6: Establishment of a reentrant arrhythmia where a Purkinje fiber (left) impinges on the ventricular myocardium near a region where decremental conduction and unidirectional block (shaded) prevents antegrade but not retrograde conduction. Although the initial impulse (dashed line labeled 1) cannot traverse the depressed region in the antegrade direction, propagation through normal tissue can allow the impulse to reach excitable tissue distal to the region of decremental conduction and unidirectional block. If the impulse is conducted slowly in a retrograde direction (dotted arrow), it can reenter the proximal myocardium region after it has recovered from the initial antegrade impulse so as to initiate a second wave of depolarization (dashed line labeled 2) that generates a premature systole. The ECG generated by these two beats is shown schematically at the bottom of the figure. |
Two phenomena, called summation and inhibition, are seen in areas of decremental conduction and unidirectional block (Fig. 16-7). Summation occurs when two subthreshold impulses that by themselves cannot initiate a response are added to generate a propagated action potential. Inhibition is seen when an action potential that enters, but cannot cross an area of decremental conduction, blocks conduction of subsequent impulses; this explains a phenomenon called concealed conduction (see below).
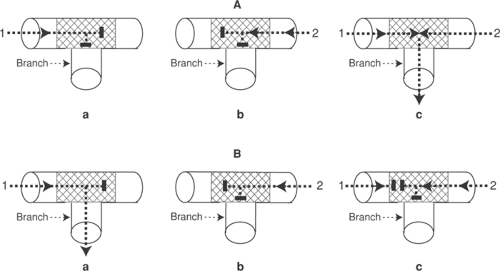 Fig. 16-7: Summation and inhibition in a branched strand of myocardium A. Summation: (a and b): Impulses 1 and 2 cannot, by themselves, generate a propagated action potential in a region of decremental conduction (cross-hatched). (c) When impulses 1 and 2 arrive simultaneously from opposite directions, they can be summated to initiate a propagated action potential in the branch. B: Inhibition: (a) When impulse 1 enters one side of a region of decremental conduction and unidirectional block (cross-hatched), it can initiate a propagated action potential. (B) When impulse 2 enters the region of decremental conduction from the other direction, its conduction is blocked. (c) When impulse 2 (which would have been blocked) enters the region of decremental conduction immediately before the arrival of impulse 1 (which would have been conducted), impulse 2 prevents impulse 1 from initiating a propagated wave of depolarization. |
Heterogeneities in Resting Potential
Localized areas of resting depolarization are a common substrate for reentrant arrhythmias, and cause many of the sudden deaths that occur during the first few minutes and hours after an acute coronary occlusion (see Chapter 17). Heterogeneous resting potential causes arrhythmias when electrotonic currents, called injury currents, that flow between partially depolarized and normal regions are able to activate resting cells (Fig. 16-8).
Heterogeneities in Action Potential Duration
Electrotonic current flow between regions of the heart that have repolarized at different rates allow inhomogeneous action potential duration to initiate tachyarrhythmias (Fig. 16-9). These inhomogeneities have many causes, including regional ischemia, which can shorten and lengthen refractoriness (see Chapter 17), end-stage heart failure, ion channel mutations, stress-activated changes in ito, and regional variations in delayed rectifier potassium channels.
Heterogeneities in Refractoriness
Disruption of the normal reactivation of depolarizing channels often occurs when abnormal refractoriness causes delayed and inhomogeneous reactivation of sodium channels, which can decrease conduction velocity and cause unidirectional block (see earlier).
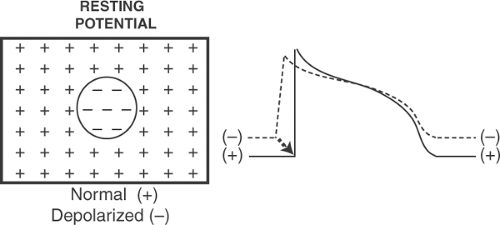 Fig. 16-8: Inhomogeneous resting potential in which the potential at the surface of a group of partially depolarized cells (center) is more negative than that of the nearby normal tissue. These differences in resting potential can allow the action potentials in the depolarized cells to activate surrounding tissue (heavy dotted arrow). |
Abnormal Conducting Structures
Abnormal conducting structures can establish electrical “short circuits” that cause reentrant tachycardias. Most common are dual AV nodal conduction pathways and accessory pathways, both of which can initiate reciprocal rhythms in which impulses traveling back and forth between the atria and ventricles cause supraventricular tachycardias.
Dual AV Nodal Conduction Pathways and AV Nodal Reentrant Tachycardias
It is not rare for the AV node to contain two pathways that conduct impulses at different velocities; the fast (more rapidly conducting) pathway, which usually lies in the lower right atrium, has a longer refractory period than the slow pathway, which is usually in the compact AV node (Fig. 16-10). AV nodal tachycardias are commonly triggered by a premature atrial
depolarization that reaches the fast pathway during its refractory period, but after the slow pathway has recovered from the preceding impulse; when this occurs, a wave of depolarization can be propagated to the AV bundle through the slow pathway after which it returns to the atria by the fast pathway (Fig. 16-10B). This can initiate a paroxysmal supraventricular tachycardia in which impulses go back and forth between the two pathways as a reciprocal rhythm.
depolarization that reaches the fast pathway during its refractory period, but after the slow pathway has recovered from the preceding impulse; when this occurs, a wave of depolarization can be propagated to the AV bundle through the slow pathway after which it returns to the atria by the fast pathway (Fig. 16-10B). This can initiate a paroxysmal supraventricular tachycardia in which impulses go back and forth between the two pathways as a reciprocal rhythm.
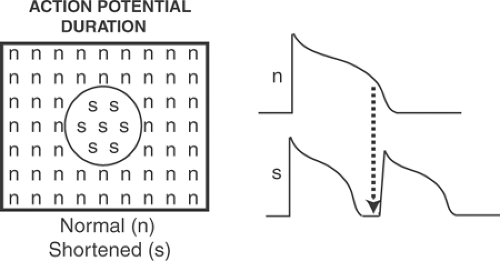 Fig. 16-9: Inhomogeneous repolarization. The early return to resting potential in a group of cells with abnormally short action potentials (s) can allow adjacent normal cells (n) to depolarize the cells with the shorter action potential (heavy dotted arrow). A similar mechanism allows cells with abnormally long action potentials to reactivate nearby normal tissue. |
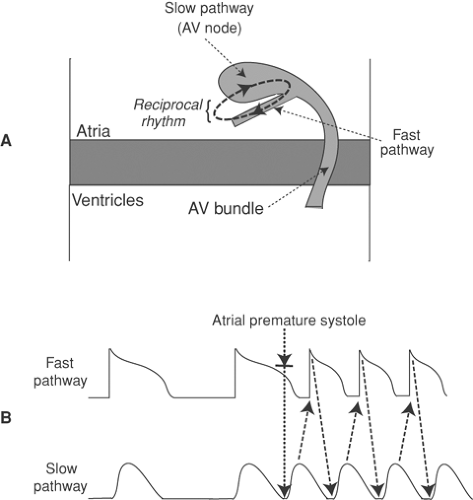 Fig. 16-10: Dual AV nodal conduction pathways. A. Anatomy. A fast conducting pathway lies in the lower right atrium, and a more slowly conducting pathway is in the atrioventricular (AV) node. The central fibrous body is depicted by the heavily shaded rectangle. B: Premature activation of the atria can initiate an impulse that travels to the ventricle via the slow pathway, which has the shorter refractory period, and then returns to the atria after reentering the fast pathway. Supraventricular tachycardias are usually caused when an atrial premature depolarization (downward dotted arrow) enters the slow pathway, but not the fast pathway (short horizontal line) because the latter has a longer refractory period. After reaching the ventricular end of the slow pathway, the premature impulse can be conducted back to the fast pathway (upward dashed arrow), and then reenter the slow pathway (downward dashed arrow). If this reciprocal rhythm continues, the reentrant circuit involving the two pathways can sustain a tachycardia. |
Accessory Pathways and Preexcitation (the Wolff–Parkinson–White Syndrome)
An accessory pathway (bundle of Kent), which is generally a strand of atrial myocardium that can be located virtually anywhere along the AV junction, can establish an electrical short circuit between the atria and ventricles (Fig. 16-11A). This pathophysiology explains the clinical features of preexcitation (the Wolff–Parkinson–White syndrome or WPW) in which conduction from the atria through the accessory pathway causes a short PR interval (<0.12 s) and distorts
the initial deflection of the QRS (called a delta wave) (Fig. 16-12). The PR interval is short because conduction through the accessory pathway is mediated by myocytes that are depolarized by fast sodium channels, and so activate the ventricles before the arrival of the impulse conducted through the AV node, where depolarization depends on the slower opening of calcium channels. The QRS complex begins abnormally (the delta wave) because impulses conducted down accessory pathways do not enter the ventricles normally via the His-Purkinje system; instead, ventricular depolarization begins at an abnormal site, which distorts the initial deflection of the QRS complex.
the initial deflection of the QRS (called a delta wave) (Fig. 16-12). The PR interval is short because conduction through the accessory pathway is mediated by myocytes that are depolarized by fast sodium channels, and so activate the ventricles before the arrival of the impulse conducted through the AV node, where depolarization depends on the slower opening of calcium channels. The QRS complex begins abnormally (the delta wave) because impulses conducted down accessory pathways do not enter the ventricles normally via the His-Purkinje system; instead, ventricular depolarization begins at an abnormal site, which distorts the initial deflection of the QRS complex.
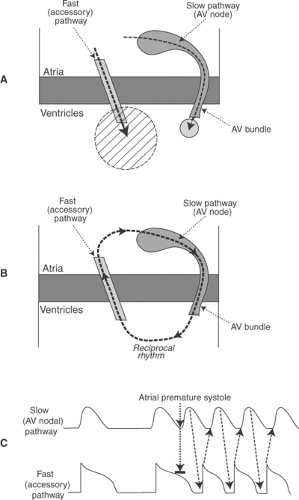 Fig. 16-11: Preexcitation. A: Anatomy. An accessory pathway (bundle of Kent), which conducts more rapidly than the pathway that includes the atrioventricular (AV) node, is usually a strand of atrial tissue that crosses the fibrous skeleton of the heart to provide an abnormal conduction pathway linking the atria and ventricles. The ventricles are activated by atrial impulses that are conducted through both pathways, but because conduction is more rapid in the accessory pathway, ventricular activation begins in an abnormal location (cross-hatched circle). The impulse conducted by the normal pathway encounters the normal delay in the AV node before it can enter the ventricles via the AV bundle (smaller shaded circle). The central fibrous body is depicted by the heavily shaded rectangle. B: Supraventricular tachycardias are usually caused by a reciprocal rhythm in which impulses travel from the atria to the ventricles via the slower AV nodal pathway, and from the ventricles to the atria via the accessory pathway. C: The reciprocal rhythm in B is initiated when an atrial premature depolarization (dotted arrow) enters the slower AV nodal pathway but not the accessory pathway because the latter has a longer refractory period. After the premature impulse reaches the ventricles (downward dashed arrow), it can return to the atria in the retrograde direction through the faster accessory pathway (upward dashed arrow), and then propagated by antegrade conduction back to the ventricles through the slower AV nodal pathway (downward dashed arrow). In this way, impulses can be conducted back and forth through the two pathways to establish a reciprocal rhythm between the atria and ventricles that causes a supraventricular tachycardia. |
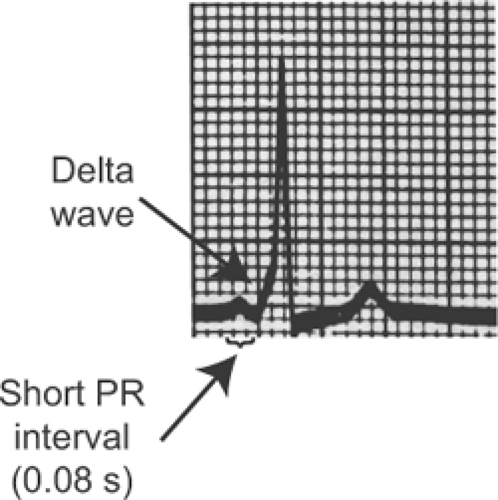 Fig. 16-12: Preexcitation (Wolff–Parkinson–White syndrome) recorded in Einthoven bipolar lead I. The PR interval is abnormally short (0.08 s) because of rapid conduction through an accessory pathway, while the slurred upstroke of the QRS complex (the delta wave) is caused when this impulse initiates ventricular depolarization at an abnormal site. |
Accessory pathways provide the substrate for supraventricular tachycardias in which impulses usually travel from the atria to the ventricles via the AV nodal pathway, and from the ventricles to the atria via the accessory pathway (Fig. 16-11B). These tachycardias begin when an atrial premature depolarization is transmitted to the ventricles only by the slower AV nodal pathway because the accessory pathway has a longer refractory period (Fig. 16-11C). After the impulse reaches the ventricles, it returns to the atria via the accessory pathway to cause a supraventricular tachycardia.
Preexcitation can occur in patients with hypertrophic cardiomyopathies caused by abnormalities in glycogen metabolism (see Chapter 18). The accessory pathways in these patients consist of myocardial strands that traverse the central fibrous body in gaps formed by glycogen accumulation.
Circus Movements
In the early 20th century, Mines used a jellyfish mantle to model reentry in a ring of excitable tissue. By gently cross-clamping the ring before stimulating the ring next to the clamp, he initiated a wave of depolarization that propagated in only one direction (clockwise in Fig. 16-13)
because conduction in the other direction was blocked by the clamp. If the clamp was removed before the impulse returned to the site of block, the wave of depolarization continued to circulate around the ring as a “circus movement” that lasted until tissue deterioration extinguished the ability to conduct (Fig. 16-14).
because conduction in the other direction was blocked by the clamp. If the clamp was removed before the impulse returned to the site of block, the wave of depolarization continued to circulate around the ring as a “circus movement” that lasted until tissue deterioration extinguished the ability to conduct (Fig. 16-14).
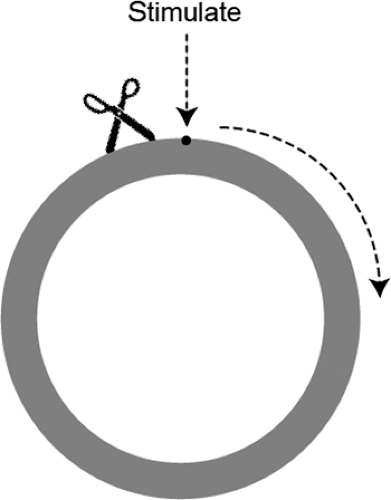 Fig. 16-13: Initiation of a circus movement. When a ring of excitable tissue is lightly clamped and stimulated alongside the clamp, counterclockwise conduction is blocked so that the wave of depolarization can propagate in only the clockwise direction. If the clamp is removed before the wave of depolarization returns to the area of block, the impulse can continue to circle the ring in a clockwise direction. |
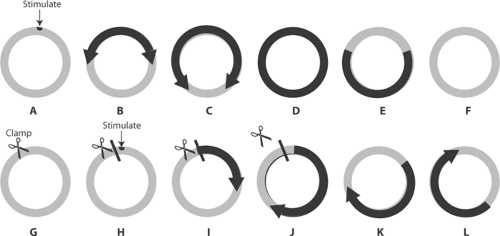 Fig. 16-14: Initiation of a circus movement in a ring of excitable tissue containing an area of decremental conduction and unidirectional block created by clamping the tissue near the site of stimulation. Resting areas are lightly shaded, and activated areas are darkly shaded. Upper row: Stimulation in the absence of block (A) depolarizes the ring in both directions (B and C) which leads to mutual cancellation of the two impulses (D) and failure to establish a circus movement (E and F). Lower row: When a clamp blocks counterclockwise propagation (G) (see Fig. 16-13), the stimulus initiates a wave of depolarization that can propagate only in the clockwise direction (H, I and J). If the clamp is removed and the impulse allowed to cross the area that had been blocked, the wave of depolarization continues to circle the ring in a clockwise direction (K and L). |
This model was used to define three mechanisms that could interrupt a circus movement (Fig. 16-15). The first is to slow conduction, for example, by inactivating the channels which carry the inward currents that propagate the wave of depolarization (Fig. 16-15a). The second is to accelerate conduction so that the wave of depolarization “catches up” with regions that are still refractory after the prior passage of the wave front (Fig. 16-15b). The third mechanism is to prolong the refractory period, which can extinguish the wave of depolarization when it reaches tissue that had not recovered from prior passage of the impulse (Fig. 16-15c). A classical analogy is to view the impulse traveling around the circular pathway as a snake which dies if its head catches up with its tail. According to this analogy, slowing conduction is like paralyzing the snake, while speeding the advance of its head or slowing the retreat of its tail allows the snake to give itself a fatal bite.
Overdrive Suppression
Most of the tachycardias listed in Table 16-2 (except for triggered activity, see below) can be terminated by stimulating the heart at a rate more rapid than the tachycardia. Several mechanisms have been proposed to explain this phenomenon, called overdrive suppression. Frequent depolarization increases sodium entry which, by elevating cytosolic sodium concentration, can facilitate sodium channel reactivation when the repolarizing current generated by the
sodium pump increases resting potential (see Chapter 8). Another explanation is that the more frequent action potentials increase calcium influx (see Chapter 10) which, by increasing energy utilization, causes energy starvation and intracellular acidosis, both of which slow conduction and so convert unidirectional block to bidirectional block.
sodium pump increases resting potential (see Chapter 8). Another explanation is that the more frequent action potentials increase calcium influx (see Chapter 10) which, by increasing energy utilization, causes energy starvation and intracellular acidosis, both of which slow conduction and so convert unidirectional block to bidirectional block.
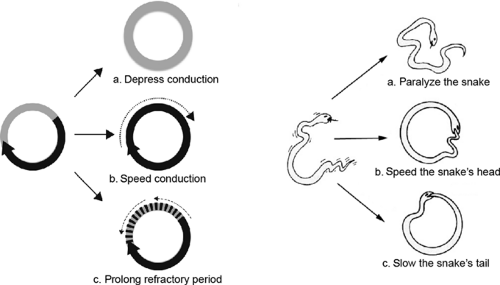 Fig. 16-15: Three ways to stop the circus movement described in Figure 16-14. Left: The circus movement can be terminated if conduction is slowed (a), accelerated (B), or if the refractory period is prolonged (c). All allow the advancing impulse to reach the previously depolarized tissue during its refractory period. Right: These mechanisms can be viewed as three ways to stop a snake from traveling in a circle. The snake stops circling if it is paralyzed (a), if its head advances fast enough to bite its tail (B), or if slowing the retreat of its tail allows the latter to come within biting distance of the advancing head (c). |
Triggered Depolarizations
Triggered depolarizations, which can result from early and late afterdepolarizations (see Chapter 14), are an important cause of premature systoles and tachycardias that can arise in the atria, His-Purkinje system, and ventricles. Because these arrhythmias are often caused by depolarizing currents generated by calcium efflux from the cytosol via the Na/Ca exchanger (see Chapter 8), triggered depolarizations occur in patients with coronary artery disease and heart failure in whom energy starvation causes cytosolic calcium overload. Rapid electrical stimulation does not suppress calcium overload-induced afterdepolarizations; instead, these arrhythmias are made worse because calcium flux into the cytosol is increased by the greater frequency of calcium channel opening.
Clinical Arrhythmias
The following pages describe the mechanisms and electrocardiographic features of several clinical arrhythmias, along with a few therapeutic principles. These examples are provided to illustrate some clinical applications of the material covered above, and in Chapters 13 to 15.
Sinus Node Abnormalities
Cardiac rate and rhythm are normally determined by depolarization of the SA (sinus) node pacemaker. By convention, a rate faster than 100 that originates in the SA node is a sinus tachycardia, while slowing of the SA node pacemaker to a rate less than 60 is a sinus bradycardia.
Sinus Tachycardia
The electrocardiographic characteristics of sinus tachycardia (Fig. 16-16) are normal QRS complexes preceded by normal P waves and normal PR intervals that occur at a rate greater than 100. P waves are normal because the tachycardia originates in the SA node and is conducted normally through the atria, while the PR interval and QRS complexes are normal because the ventricles are depolarized normally via the AV bundle and bundle braches. (When the heart is diseased or heart rate is extremely rapid, atrioventricular and intraventricular conduction can be abnormal in sinus and other supraventricular tachycardias.) Sinus tachycardias typically begin and end gradually, and are slowed by vagal stimulation; these characteristics distinguish this common arrhythmia from most other supraventricular tachycardias, which start and stop suddenly (see below).
Increased sympathetic activity and reduced vagal tone, which occur normally during physical exercise and emotional stress, are the most common causes of sinus tachycardia (see Chapter 14); other causes include fever, anemia, hyperthyroidism, and stretch of the SA node (the Bainbridge reflex). The maximum rate of normal exercise-induced sinus tachycardia is sometimes stated to be 220 minus the patient’s age, so that a healthy 20-year-old should be able to achieve a sinus rate of 200, while maximum sinus rate in a 70-year-old should be only 150. This relationship, however, is highly variable.
Sinus Bradycardia
Sinus bradycardia occurs when SA node pacemaker rate is less than 60. The ECG in sinus bradycardia therefore shows a slow heart rate in which normal P waves are followed by normal QRS complexes after a normal PR interval (Fig. 16-17). The most common causes of sinus bradycardia are increased parasympathetic (vagal) activity and decreased sympathetic activity (see Chapter 14). Physical conditioning increases the tonic parasympathetic tone that normally slows the sinus pacemaker, which explains the resting sinus bradycardia characteristic of the “athlete’s heart.” Severe sinus bradycardia can cause vasovagal syncope (the “swoon” in 19th-century novels), and can be triggered by reflexes in patients with acute inferior or posterior myocardial infarction (see Chapter 17). Drugs that cause sinus bradycardia include β-adrenergic receptor blockers and some L-type calcium channel blockers. The former inhibit pacemaker
activity indirectly when reduced synthesis of cyclic AMP decreases PKA-catalyzed phosphorylation of the channels responsible for several pacemaker currents (see Chapter 14), while the latter act directly to inhibit calcium channel opening in the SA node. Cardiac glycosides slow the SA node by a central effect that increases parasympathetic activity.
activity indirectly when reduced synthesis of cyclic AMP decreases PKA-catalyzed phosphorylation of the channels responsible for several pacemaker currents (see Chapter 14), while the latter act directly to inhibit calcium channel opening in the SA node. Cardiac glycosides slow the SA node by a central effect that increases parasympathetic activity.
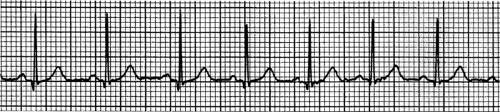 Fig. 16-16: Sinus tachycardia (lead II). The rates of atrial and ventricular depolarization are approximately 120. Each QRS complex, which is normal, follows a normal P-wave after a normal PR interval of 0.12 s (retouched). |
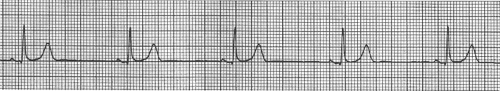 Fig. 16-17: Sinus bradycardia (lead II). Normal QRS complexes follow normal P-waves after a normal PR interval, but at a rate of 45. |
Sinus Arrhythmia and Wandering Pacemaker
A perfectly regular sinus rhythm is not, as might be expected, a sign of a healthy heart; instead, small variations in cycle length, called sinus arrhythmia, are characteristically seen in normal individuals. This variability, which is due largely to physiological changes in sympathetic and parasympathetic tone, is lost in patients with heart disease, in part because of sustained over-stimulation of the neurohumoral response (see Chapter 8). For this reason, clocklike regularity of the heartbeat suggests that the normal fine tuning by the autonomic nervous system has been blunted. Decreased heart rate variability is one of the first signs of heart disease, so that sinus arrhythmia is generally a sign of good health!
Changes in sinus rate that accompany normal respiration, called respiratory sinus arrhythmia, alter cycle length without affecting impulse propagation through the atria, AV node, and ventricles; for this reason, the only changes are in the firing rate of the SA node (Fig. 16-18). Respiratory sinus
arrhythmia occurs when stimuli arising in stretch receptors in the lung and chest wall alter the balance between sympathetic and parasympathetic influences on the SA node; heart rate accelerates during inspiration because of decreased vagal tone and increased sympathetic tone, while the SA node pacemaker slows when expiration increases vagal tone and decreases sympathetic tone.
arrhythmia occurs when stimuli arising in stretch receptors in the lung and chest wall alter the balance between sympathetic and parasympathetic influences on the SA node; heart rate accelerates during inspiration because of decreased vagal tone and increased sympathetic tone, while the SA node pacemaker slows when expiration increases vagal tone and decreases sympathetic tone.
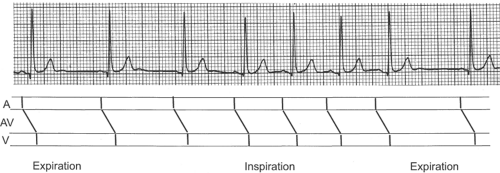 Fig. 16-18: Respiratory sinus arrhythmia (lead II). Atrial and ventricular depolarization accelerate during inspiration and slow during expiration, but each QRS complex is preceded by a normal P wave and a normal PR interval. The arrhythmia is diagrammed below, using a “Lewis diagram”; the eponym recognizes Sir Thomas Lewis, a pioneer in electrocardiography. The top line represents the sinoatrial (SA) node, the upper space (A) the atria, the middle space (AV) the AV node, and the lower space (V) the ventricles. The downward angle of the lines represents the speed of impulse propagation. Each beat in this arrhythmia begins in the SA node and is conducted normally through the atria, AV node, and ventricles. |
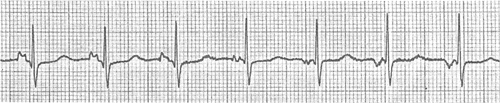 Fig. 16-19: Wandering pacemaker (lead II). The rate in this series of sinus beats is regular, with cycle lengths between 0.69 and 0.72 s. The marked changes in P wave morphology can be explained by shifts in the location of the pacemaker within the sinoatrial node. |
Another common benign SA node arrhythmia is wandering pacemaker, which is characterized by changes in P wave morphology and minor variations in cycle length (Fig. 16-19). This arrhythmia, which can be related to respiration, is caused by shifts in the site of pacemaker activity within the SA node (see Chapter 15).
Sinoatrial Block
Heart rate can be slowed when impulses arising in the SA node fail to depolarize the atria (Fig. 16-20). This abnormality, called SA block, can be difficult to distinguish from sinus bradycardia because depolarization of the SA node pacemaker cannot be recorded by the ECG (see Chapter 15). SA block can be caused by increased vagal tone, drugs, and other factors that decrease depolarizing currents in the SA node pacemaker (see above). The latter include occlusion of the SA node artery, apoptosis, fibrosis, and degeneration of the nodal cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


