Chapter 46 Approach to Diagnosis of Diffuse Lung Disease
• Cryptogenic organizing pneumonia (bronchiolitis obliterans organizing pneumonia, proliferative bronchiolitis)
• Idiopathic pulmonary fibrosis (cryptogenic fibrosing alveolitis)
• Hypersensitivity pneumonitis (extrinsic allergic alveolitis)
This problem has been partially addressed by the reclassification of the idiopathic interstitial pneumonias by a joint American Thoracic Society/European Respiratory Society (ATS/ERS) international consensus committee, discussed in detail in Chapter 47. However, the term cryptogenic fibrosing alveolitis (CFA) continues to cause difficulties. As defined in the ATS/ERS reclassification, CFA is strictly synonymous with idiopathic pulmonary fibrosis (IPF). The diagnosis of IPF/CFA now requires the presence of usual interstitial pneumonia (UIP) at surgical biopsy or typical appearances on HRCT, in association with a compatible clinical picture. This represents a radical change; in historical series, various disorders presenting with a clinical picture of IPF were grouped together as IPF/CFA. The entity of “clinical CFA syndrome” is still necessary for epidemiologic studies but should not be viewed as a final diagnosis in clinical practice.
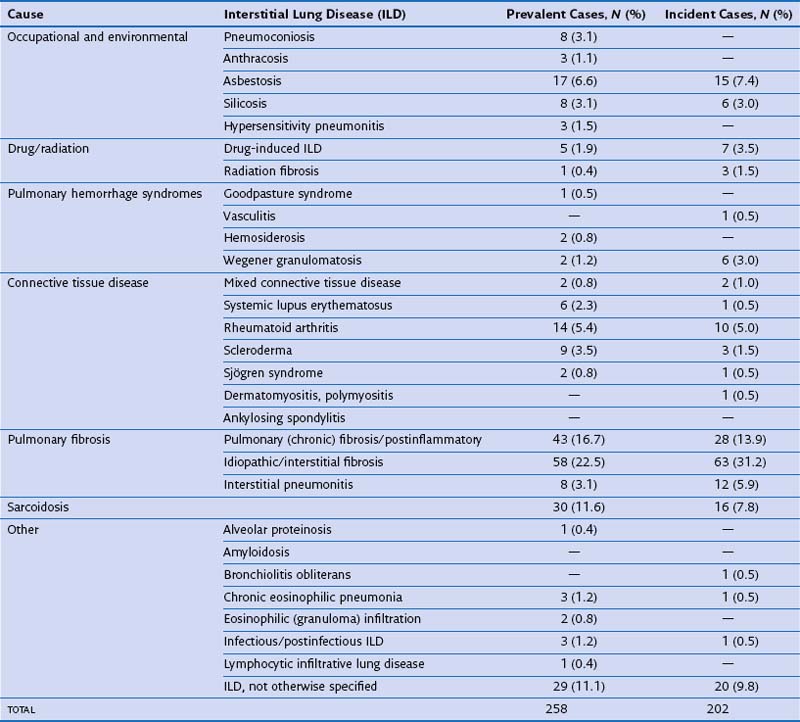
In routine practice, a simplified pragmatic approach to diagnosis of DLD is essential; consideration of a checklist of the more common diseases is often useful. The classification of DLD by their disease burden was addressed most definitively in a study from Bernalillo County, New Mexico, in which the incidence and prevalence of individual DLDs was quantified using a variety of methods (Table 46-1). New cases were estimated to occur in 32 : 100,000 years in males and 26 : 100,000 years in females; thus, although less common than lung infection, malignant disease or obstructive airways disease, the DLDs are responsible for a considerable disease burden. More recent evidence shows an increase in the prevalence of DLD, especially IPF, which inevitably means that the burden of disease has increased further in the previous one to two decades. Moreover, the workload for the respiratory medicine physician is disproportionate because the diagnosis of individual DLDs is often uncertain, despite more intensive investigation than is generally required in obstructive airways disease, malignancy, or chronic lung suppuration.
Initial Clinical Evaluation
Clinical History
The identification of an underlying cause is the single most important contribution made by clinical evaluation. Table 46-2 provides a checklist of the more important causes of DLD. A careful occupational history is essential and should include details of all previous occupations, including short-term employment. Asbestos exposure is often extensive in railway rolling-stock construction, shipyard workers, power station construction and maintenance workers, naval boilermen, garage workers (involved in brake lining), and other occupations in which asbestos exposure is overt; generally, workers in these occupations are well aware of their asbestos exposure. However, other workers, including joiners, electricians, carpenters, and construction workers, who handle asbestos in the form of roofing and insulation material, are often unaware of significant exposure. Other occupations associated with DLD include coal mining (coal worker’s pneumoconiosis), metal polishing (hard metal disease), and sandblasting (silicosis).
Table 46-2 Frequently Encountered Diffuse Lung Diseases with Identifiable Underlying Cause
| Cause | Differential Diagnosis |
|---|---|
| Occupational-related or other inhalant–related | |
| Inorganic | |
| Organic | |
| Collagen vascular disease–related | |
| Drug-related | |
| Physical agents/toxins | |
| Neoplastic disease | |
| Vasculitis-related | |
| Disorders of circulation | |
| Chronic infection | |
| Smoking-induced | |
A detailed drug history is also essential. The drugs most frequently causing DLD are probably amiodarone, methotrexate (at doses used in CTD), and antineoplastic agents, especially bleomycin. However, a wide variety of other agents (>200 at present) cause DLD, although often in only a small number of patients, and the list increases yearly. Fortunately, an international website is now devoted to drug-induced lung disease (www.pneumotox.com), through which all medications should be routinely checked in patients with DLD.



