Applications of Proteomics to the Management of Lung Cancer
Applications of Proteomics to the Management of Lung Cancer
Pierre P. Massion
David P. Carbone
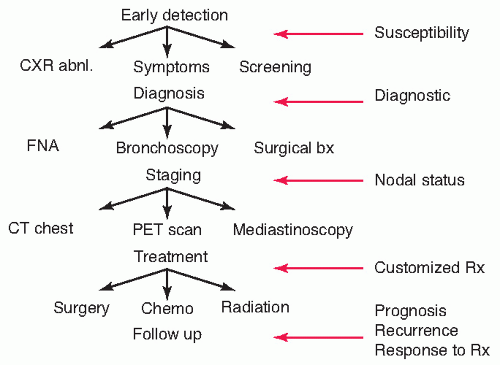
Lung cancer is the third most common cancer in the United States, yet it causes more deaths than breast, colon, pancreas, and prostate cancer combined.1 Attempts to alter these statistics have been challenging in all respects. From risk assessment to diagnosis, staging, assessment of response to therapy, and prognostication, there is much room for improvement in this disease (Fig. 9.1). Rapid developments in technology have allowed the direct analysis of patterns of protein expression in tumors and blood samples, and these proteomic analyses promise to assist in making progress in each of these areas.
Proteins are ultimately responsible for the function of the vast majority of biological systems, and it is clear that many of the crucial proteins in a cell are primarily regulated by posttranslational modifications such as proteolytic processing, phosphorylation, or acetylation. Thus, a full knowledge of the derangement in the expression, modification, and function of proteins in cancer cells is likely to be more informative than study of DNA or RNA alone. The aim of proteomics is therefore the characterization of proteins to obtain a more integrated view of the biology. In order to further understand the molecular biology of lung cancer, we need to probe these tissues and related biological materials with tools that address the molecular complexity of the proteome in lung cancer. New technologies are being rapidly developed to allow the increasingly thorough, systematic, and simultaneous analyses of thousands of proteins in cancer cells. In particular, these studies give us a unique insight into the biology of cancer, can yield important new therapeutic targets, and may enable the identification of novel biomarkers to differentiate tumor from normal cells and predict individuals likely to develop lung cancer.
In this chapter, we will review the progress made in clinical proteomics as it applies to the management of lung cancer. We will focus our discussion on how this approach may advance the areas of early detection, response to therapy, and prognostic evaluation.
PROTEOMICS TECHNOLOGIES
Sample Preparation With proteomics strategies, one strives to identify novel proteins and understand their structure, function, interaction with proteins and other molecules, and to bring this knowledge to the clinic by means of new diagnostic and predictive biomarkers and well as identification of therapeutic targets. The rapid advance of mass spectrometry (MS) and related technologies offers powerful new tools to analyze the proteome. In contrast to standard protein biochemistry, proteomics is defined as the study of the proteome, the complete set of proteins produced by a species, using the technologies of large-scale protein separation and identification (Table 9.1).2
Protein biochemistry has long been exploited to understand how biological systems function and lead to a cancer phenotype. Early efforts were geared to study primarily one protein at a time with biochemical methods of increasing power and sensitivity. The world of immunoassays brought and continues to bring major contributions to the field alone or in combination with MS.
In proteomic analysis, the isolation and preparation of samples for analysis is of critical importance, and the precise technique chosen depends on the scientific question being addressed, whether it may be a comprehensive expression analysis, evaluation of secreted proteins, nuclear proteins, proteins with a particular modification (e.g., phosphorylation), or those that bind to other proteins. Many approaches are available: some gel-based, some based on separation by reverse phase chromatography, affinity, size exclusion, ion exchange, and isoelectric focusing. The separation/purification strategies all have the advantage of separating the targets of interest from very abundant proteins in the milieu. The trade-off is between the addition of complexity and variability to the analysis and increased sensitivity. For example, proteomics of blood samples is complicated by the fact that the vast majority of the proteins in blood are made up of albumin and immunoglobulin, but the proteins of interest may be 7 to 10 orders of magnitude less abundant, and much more readily found if the abundant proteins are removed. An example of successful separation strategy is immunoaffinity phosphoproteomics, which combines immunoaffinity purification with tandem MS. Using this approach, Rikova et al.3 discovered oncogenic kinases such as platelet-derived growth factor receptor (PDGFR)-alpha and discoidin domain receptor family, member 1 (DDR1) that had not been implicated in the pathogenesis of lung cancer.
FIGURE 9.1 Schematic representation of major areas of management of lung cancer in the clinical context and potential applications of proteomic approaches to address questions of susceptibility, diagnosis, staging, and therapeutics. abnl, abnormal; bx, biopsy; CT, computed tomography; CXR, chest x-ray; FNA, fine-needle aspiration.
The Mass Spectrometer A mass spectrometer analyzes proteins after their conversion to gaseous ions, based on their mass to charge ratio. It is essentially made of three basic elements: an ion source for converting them to gaseous ions, a mass analyzer for separating the ions by mass, and a detector for detecting the ionized proteins (Fig. 9.2).
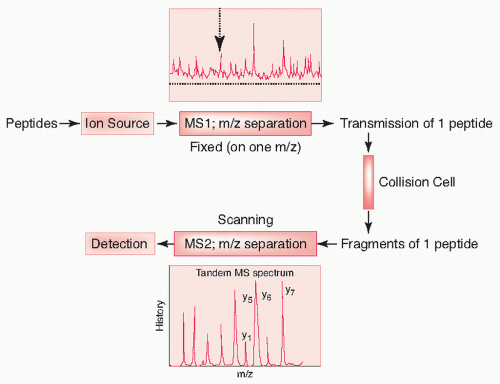
Tandem mass spectrometry (MS/MS) is a major analytic tool used for evaluating proteins and protein complexes. With this approach, protein samples are first digested with proteases into a mixture of peptides and analyzed. Peptide ions are separated in the first stage, then each peptide is fragmented in the collision cell, and the fragments are then separated again to identify them. The precise measurement of the mass of these fragments allows the reconstruction of the identity and composition of the original peptide (Fig. 9.3). There are many modes of MS/MS. Different mass analyzers currently used for MS/MS analysis are quadrupole ion trap (QIT), triple quadrupole (TQ), quadrupole time of flight (QTOF), or Fourier transform ion-cyclotron resonance (FTICR).
FIGURE 9.2 Principles of mass spectrometry analysis. The time-of-flight (TOF) analyzer uses an electric field to accelerate the ions, and then measures the time they take to reach the detector. A quadrupole mass analyzer acts as a mass-selective filter. The quadrupole ion trap works on the same physical principles as the quadrupole mass analyzer, but the ions are trapped and sequentially ejected. BAL, bronchoalveolar lavage; FTICR, Fourier transform ion-cyclotron resonance; LTQ, linear quadrupole ion trap; MALDI, matrix-assisted laser desorption ionization; MS, spectometry. The Orbitrap is a novel form of MS instrumentation with 60 to 100 k resolution and <2 ppm mass accuracy.
TABLE 9.1 Analytical Approaches to the Proteome
Protein biochemistry
Immunoblotting
Immunohistochemistry
ELISA
Flow cytometry
Protein chemical analysis
Antibody production
Proteomics
Sample preparation for isolation of proteins
Gel-based separation followed by MS
LC-HPLC
Affinity columns
Affinity tags
Mass spectrometry-based protein identification
MALDI MS/MS
ESI MS
Electron transfer dissociation MS
Protein arrays including reverse phase arrays
Specific mass spectrometers have specific applications. For example, electron transfer dissociation (ETD) MS allows detailed analysis of phosphorylated peptides that is of optimal quality.4 The LTQ integrates the steps of mass analyzer, collision cell, and then another mass analyzer actually does it tandem in time, meaning that three steps occur in the same location—in an ion trap. An LTQ tandem MS is particularly well equipped for excellent throughput, good sensitivity, MSn capabilities, and a robust instrument. Each clinical question needs to be addressed separately and proteomics may provide tools to address them. The suite of technologies available to researchers is ever increasing and their selection very much depends on the goals and the type of samples to analyze.
FIGURE 9.3 Principle of tandem mass spectrometry. A peptide mixture is injected into the MS. Unlike the singlestage mass spectrometer that scans the entire range of masses, the first analyzer is set up to transmit only one peptide —therefore, one mass over charge (m/z). This peptide is then broken apart in a collision cell to generate peptide fragments. The m/z of these peptides is then determined by another mass analyzer. The amino acid sequence of the peptide is determined by subtracting fragment ion masses from each other, yielding the residue mass of a particular amino acid. The process is repeated until the sequence has been resolved.
Analysis of Complex Protein Mixtures The proteome has multiple layers of complexity. The composition of the proteome is not static like DNA. The structure is at least an order of magnitude more complex than the genome, the dynamic range in protein concentrations in given biological specimens is huge (10 12), we have no target amplification method such as PCR for genomic analysis, and the methods used still have limitations in sensitivity primarily because of our ability to separate protein complexes in subgroups pure enough for analysis. Quantitative analysis is also challenging, but new methodologies are being developed to address this challenge. High-throughput analysis of the proteome without compromising reproducibility is difficult as well.
The analysis of the complex mixture can be conceptualized in two major ways, the top-down and the bottom-up approaches. In the top-down approach, one starts with a specific protein candidate; it is then separated, purified, and its structure is identified. Recent technologies, such as FITRC MS, increase the resolution and allow the analysis of larger peptide fragments. However, the bottom-up approach takes the challenge of embracing complexity from the start and directly analyzes complex mixtures with a large number of proteins, and uses computational peptidomics to reconstruct the identities of the proteins in the mixture. This later approach is less intuitive and may benefit from higher throughput. It is recently being facilitated by modern bioinformatics tools, enabling the analysis of proteomic digestion with different enzymes than trypsin and therefore increasing the likelihood of detecting increasing number of peptides mapping to the same protein therefore improving the confidence of identification.
Biomarkers The assumption underlying the concept of proteomic biomarkers is that certain characteristics of proteomes are highly correlated with specific clinically relevant biological states. These characteristics include changes in expression levels of proteins and the presence of specific modified protein forms (Table 9.2). Specific effort has been exerted in cancer proteomics to develop biomarkers for the early detection of disease by analysis of plasma or serum proteins. Detection of cancers at early stages maximizes survival, and identification of blood-borne markers would lead to minimally invasive tests.
The best biomarkers are those that are reproducibly measured, related to the disease process, and trigger a clinical decision resulting in improved clinical outcomes. Despite an intense search for such biomarkers in the last 20 years, there are none currently available for early diagnosis of lung cancer.5 One reason for such a lack of success thus far is the enormous challenge offered by lung cancer development. The onset of the disease process is extremely slow (months to years) and we have no means of evaluating the rate of progression. Therefore, there is a critical need for new biomarkers that are related to the disease process and that can be measured early, easily, and repeatedly to assess progression of the process.
TABLE 9.2 Characteristics of a Biomarker and the Process Selection of Candidates
Characteristics of a Biomarker
Should be an indicator or surrogate marker of clinical end point
Should be measurable, quantifiable, reproducible
Should evaluate a biological process and predict the outcome
Process of Selection of Candidates
Demonstrate that marker appears in accessible material
Establish quantitative criteria for the presence of the marker
Validate marker against accepted end points
Confirm its predictive value in prospective study
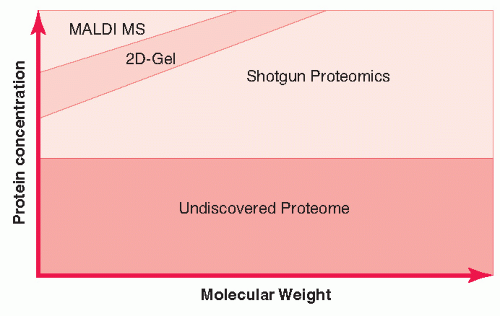
Approaches to Biomarker Discovery Using Proteomics Biomarker identification has been addressed by multiple proteomic technologies (Fig. 9.4). MALDI profiling is rapid, high throughput, but detects only the most abundant proteins of relatively low molecular weight, and does not enable direct identification when applied to complex proteomes. Two-dimensional (2D) gel-based analysis suffers problems of interlaboratory reproducibility and throughput. More recent in-depth proteomic analyses are trying to overcome these limitations and are summarized here.
High-Throughput Profiling Techniques The rapid proteomic profiling of blood, tissue, or urine with minimal sample preparation, using the peak pattern as a diagnostic tool, has generated great enthusiasm and yet has been minimally successful at providing robust signatures to translate to the clinic. In this approach, the focus is on the use of MS peak patterns of abundant proteins or peptide fragments that correlate with an early disease stage but are usually not part of the disease mechanism. MALDI TOF MS is capable for very high throughput where a sample can be analyzed in seconds and has higher tolerance for salts, buffers, and other biological contaminants. Because of these qualities, MALDI MS has been utilized to study proteins/peptides in serum,6,7,8,9,10 urine,11 tissue extracts,12,13 whole cells,14 and laser-captured microdissected cells.15
FIGURE 9.4 Proteome and analytical coverage of its complexity by current technologies according to protein concentration (depth) and molecular weight (breath). MALDI MS, matrix-assisted laser desorption ionization mass spectometry.
Profiling using this technology in biological fluids or tissue samples is not without challenges. The enormous complexity of the sample composition, the large dominance of few proteins in the sample, and their ability to mask lower abundance limits the informativity of this approach. Truly tumor-derived markers are likely to be present at low levels in blood, similar to levels of the thousands of other proteins in blood that derive from normal tissue leakage. Thus, the dynamic range of protein concentrations adds a new dimension of technical considerations to successful analysis of the serum/plasma proteome.16 These profiling experiments have been applied to a series of biological specimens. Yet, reproducibility between platforms and institutions remains a problem such that none of the profiling experiments have yet made an impact in clinical medicine. This is in contrast with greater early steps in the translation of genomic signatures to the clinic.17,18
Finally, protein arrays have recently been developed and offer a series of targets printed onto different surfaces. Proteins,19 peptides, antibodies,20,21 or lysates22 will then be detected by antibodies, serum, or multicolor detection systems. The Swedish Human Protein Atlas (HPA) program proposes a systematic analysis of the human proteome using antibodybased proteomics combining affinity-purified antibodies with protein profiling assembled in tissue microarrays.23
In-Depth Proteomics Analysis The analysis of the plasma proteome has made great progress in the last few years. http://www.hupo.org/research/hppp/. This is largely a consequence of novel methods of serum fractionation and MS-based protein identification techniques; the number of plasma proteins now includes major categories of proteins in the human proteome.24 The list confirms the presence of a number of interesting candidate marker proteins in plasma and serum.25 The detection of low-abundance proteins in the plasma requires combinations of powerful technologies. The identification of proteins whose expression levels are altered with the disease state progression (2DE, MS, shotgun proteomics) requires methodological improvements over the profiling experiments. Methods related to separation of ions and ionization have moved the field forward.
Two technology platforms have been developed to enable unbiased discovery of candidate markers from tissues and biofluids and verification of candidate markers by targeted analysis. Unbiased discovery employs a shotgun proteomics platform based on isoelectric focusing of peptides from tissue protein digests, followed by reverse phase LC-MS-MS on Thermo LTQ or LTQ-Orbitrap instruments. Verification is done by targeted quantitation of peptides derived from biomarker candidate proteins using liquid chromatography—multiple-reaction monitoring MS (LC-MRM-MS).26,27,28
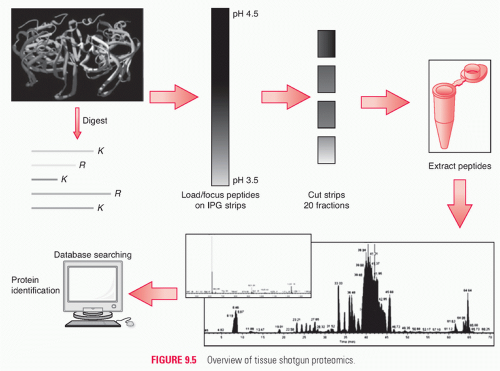
In shotgun analyses, protein mixtures are digested to peptides, which then are analyzed, most commonly by multidimensional LC-MS-MS. MS-MS spectra encode the sequences of peptides, as well as the masses and sequence positions of any modifications (Fig. 9.5). Matching of MS-MS spectra to database sequences enables identification of the peptides and the proteins from which they were derived. Shotgun analyses by LC-MS-MS also result in direct identification of the peptides detected and provide for quantitative analysis of protein components. Shotgun proteomics has proven the most versatile and effective method for dissecting multiprotein complexes, signaling networks, and complex subcellular proteomes.29 Shotgun analyses can confidently identify 3000 to 5000 proteins from a 200-μg protein sample. Shotgun analyses have generated the most complete proteomic inventories to date of major eukaryotic subcellular organelles, whole cell and tissue proteomes, and proteomes of human biofluids, including plasma and serum.28,30,31,32,33
FIGURE 9.5 Overview of tissue shotgun proteomics.
Targeted quantitative analysis of top candidates can be done by LC-MRM-MS analysis, as a first level of verification of the shotgun results. Briefly, tissue lysates are run on a NuPAGE gel and peptides are extracted, then injected in a TSQ Quantum Ultra mass spectrometer. Peptides are loaded and desalted and resolved in reverse phase chromatography, eluted with a linear gradient. For MRM, four transitions are recorded, and chromatographic peak areas for the transitions are summed and compared to summed peak areas for beta actin. Differences between peaks are evaluated for statistical significance. Targeted LC-MRM-MS analyses can analyze up to 100 candidates per run in individual tissue or plasma specimens. Moreover, the application of both stable isotope tagging and label-free quantitation has enabled the application of shotgun proteomics to quantitative comparisons of complex proteome samples.34,35,36,37,38,39
Only gold members can continue reading. Log In or Register to continue