Aortic Stenosis
Joshua M. Friedland-Little
Jeffrey D. Zampi
Robert J. Gajarski
Introduction
Congenital obstruction of the left ventricular outflow tract comprises a heterogeneous group of disorders, with obstruction potentially occurring below, above, or at the level of the aortic valve. Each of these scenarios represents a distinct disease process with unique ontogeny and natural history. At the same time, there are also common themes in pathophysiology, presentation, and evaluation shared between entities. This chapter will provide an overview of left ventricular outflow tract obstruction in pediatric patients with two-ventricle physiology. Hypoplastic left heart syndrome and its variants are discussed separately (see Chapter 46).
Epidemiology
Valvar aortic stenosis constitutes the most common type of congenital left ventricular outflow tract obstruction, accounting for approximately 80% to 85% of cases (1). Structural abnormalities of the aortic valve range from potentially asymptomatic malformations (bicuspid aortic valve) to severe, ductal-dependent lesions (critical aortic stenosis), and when grouped together these anomalies constitute the most common class of congenital heart disease. A bicuspid aortic valve has been identified in approximately 1% of the general population in autopsy studies (2,3) as well as large scale echocardiography screenings of healthy, asymptomatic individuals (4). Clinically significant valvar aortic stenosis is much less common with a reported incidence between 0.2 and 0.4 cases per 1,000 live births (5,6). Severe aortic stenosis presenting in infancy comprises approximately 10% of the total cases (7). Congenital aortic valve disease occurs primarily in males; males account for 65% of cases of bicuspid aortic valve (8) and 70% to 80% of cases of congenital valvar aortic stenosis (1,9). The risk of aortic valve disease may also be modified by race, with African Americans appearing to have a lower prevalence of both bicuspid aortic valve (10) and severe aortic stenosis compared to Caucasians (11).
The second most common type of left ventricular outflow tract obstruction in children is fixed subvalvar stenosis, which accounts for approximately 15% of total cases (1,12). As with valvar aortic stenosis, subvalvar stenosis is more common in males, with a male-to-female ratio between 1.5:1 and 2.5:1 (13,14). The final broad category of aortic stenosis, supravalvar stenosis, is the least common, occurring in approximately 1 in 20,000 live births (15). Supravalvar aortic stenosis is often associated with Williams–Beuren syndrome, although it may also occur independently in both familial and sporadic patterns. Although Williams–Beuren syndrome is known to affect males and females equally, most published series of patients with supravalvar aortic stenosis have demonstrated a slight male predominance (52% to 55%) (16,17). Though the mechanism is unclear, there is evidence that among patients with Williams–Beuren syndrome, both the prevalence and severity of supravalvar aortic stenosis are increased among males (18).
Embryology and Pathology
Valvar Aortic Stenosis
Morphogenesis of the aortic valve in humans begins at approximately day 30 of embryonic development shortly after the primitive heart tube has undergone looping (19). The initial step in aortic valve development is the increased production of extracellular matrix (primarily proteoglycans and hyaluronan) by myocardial cells lining the developing outflow tract. The deposition of this extracellular matrix creates endothelial-lined “swellings” within the outflow tract. A subpopulation of the endothelial cells lining these swellings is induced to undergo the process of endothelial-to-mesenchymal transformation (EMT), after which the new mesenchymal cells migrate into and populate the collection of extracellular matrix (19,20). The cytokine vascular endothelial growth factor (VEGF) plays a key role in regulating endothelial cell proliferation to maintain a population of cells capable of undergoing EMT in the outflow tract cushions. Additional participants in the EMT process include transforming growth factor-β (TGF-β), bone morphogenetic protein 2 (BMP2), as well as the Wnt and Notch signaling pathways (19). Neural crest cells are also thought to contribute to outflow tract endocardial cushion formation and semilunar valve morphogenesis (21,22).
After the outflow tract endocardial cushions have formed, they fuse together and elongate to create the primordial aortic and pulmonary valves. As the valve interstitial cells begin to differentiate, they lose expression of mesenchymal markers and begin to express markers more characteristic of connective tissue cells (19). The differentiated cells become progressively less proliferative through embryogenesis, and postnatally there is minimal proliferation within the valve leaflets. The extracellular matrix in the developing valve leaflets also undergoes extensive remodeling, with increased deposition of collagen and proteoglycans. Elastin (ELN) deposition in the valve leaflets does not generally occur until after birth (23).
The mature aortic valve is comprised of three valve leaflets, or cusps, and three commissures (Fig. 44.1). The valve is surrounded by a fibrous ring, or annulus, to which the cusps are attached. Each aortic valve cusp is traditionally referred to according to its corresponding sinus of Valsalva, that is, as the right-coronary, left-coronary, or noncoronary artery cusp (Fig. 44.2, Video 44.1). Autopsy studies have revealed that while cusps are generally similar in size to each other, there is often minor variation in cusp size within a “normal” valve (24). Each aortic valve cusp is composed of three layers of extracellular matrix surrounded by a thin layer of valve endothelial cells (19). The first layer beneath the endothelium on the aortic surface of the valve is the fibrosa, which is comprised of fibroblasts and circumferentially arranged collagen fibers. Immediately beneath the endothelium on the ventricular surface of the valve is the ventricularis, composed primarily of ELN fibers in a radial arrangement. In between the fibrosa and the ventricularis is the spongiosa, a compressible layer of fibroblasts, proteoglycans, and mesenchymal cells.
By far the most common congenital abnormality of the aortic valve is partial or complete fusion of two of the valve cusps,
resulting in a bicuspid rather than tricuspid valve. There is often a fibrous ridge, or raphe, at the site of fusion. In 95% of cases, the cusps of a bicuspid valve are unequal in size (25), with a raphe frequently present in the larger, fused cusp (Fig. 44.3, Video 44.2). In most instances (70% to 85%) fusion occurs between the right- and left-coronary cusps (8,25,26). Fusion between the right and the noncoronary cusp is next most common, while fusion between the left and the noncoronary cusp is quite rare. A bicuspid valve is most often hemodynamically insignificant at birth, and only ∼2% of bicuspid valves develop clinically significant stenosis or insufficiency by adolescence (27). The development of clinical disease is correlated with valve cusp morphology, with fusion of the right and noncoronary cusps entailing more than twice the risk of significant aortic stenosis or insufficiency compared to the more common intercoronary cusp fusion (8).
resulting in a bicuspid rather than tricuspid valve. There is often a fibrous ridge, or raphe, at the site of fusion. In 95% of cases, the cusps of a bicuspid valve are unequal in size (25), with a raphe frequently present in the larger, fused cusp (Fig. 44.3, Video 44.2). In most instances (70% to 85%) fusion occurs between the right- and left-coronary cusps (8,25,26). Fusion between the right and the noncoronary cusp is next most common, while fusion between the left and the noncoronary cusp is quite rare. A bicuspid valve is most often hemodynamically insignificant at birth, and only ∼2% of bicuspid valves develop clinically significant stenosis or insufficiency by adolescence (27). The development of clinical disease is correlated with valve cusp morphology, with fusion of the right and noncoronary cusps entailing more than twice the risk of significant aortic stenosis or insufficiency compared to the more common intercoronary cusp fusion (8).
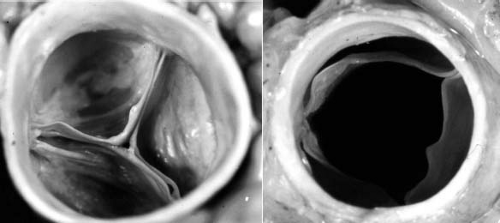 Figure 44.1 Pathologic specimen of normal aortic valve viewed from the aortic side in diastole (A) and systole (B). |
Much less common than a bicuspid aortic valve is a unicuspid aortic valve, characterized by complete or partial fusion of two (unicuspid, unicommissural) or all three commissures (unicuspid, acommissural) (28). In cases of a unicuspid, unicommissural valve, a single, posteriorly oriented commissure is most commonly seen (Fig. 44.4) (29). Not surprisingly, given the more abnormal valve morphology, unicuspid aortic valves tend to develop clinically significant disease earlier in life compared with bicuspid valves (30) and are often seen in cases of severe, ductal-dependent aortic stenosis during the neonatal period. Tricuspid aortic valves with dysplastic leaflets can also
develop significant stenosis in childhood, though less frequently than bicuspid or unicuspid valves. Regardless of cusp number, valvar aortic stenosis is generally due to incomplete opening of a “doming” valve resulting in a reduced effective valve orifice (Fig. 44.5, Video 44.3). Much less commonly, stenosis can result from annular hypoplasia. This is generally seen in the setting of additional left-sided anomalies along the spectrum of hypoplastic left heart syndrome. Calcific aortic stenosis, of either bicuspid or tricuspid aortic valves, is a common disease in adulthood but rarely seen in pediatric patients. Calcification of a bicuspid aortic valve does not generally begin until the fourth decade of life, while sclerosis can be seen as early as the second decade in otherwise asymptomatic valves (31).
develop significant stenosis in childhood, though less frequently than bicuspid or unicuspid valves. Regardless of cusp number, valvar aortic stenosis is generally due to incomplete opening of a “doming” valve resulting in a reduced effective valve orifice (Fig. 44.5, Video 44.3). Much less commonly, stenosis can result from annular hypoplasia. This is generally seen in the setting of additional left-sided anomalies along the spectrum of hypoplastic left heart syndrome. Calcific aortic stenosis, of either bicuspid or tricuspid aortic valves, is a common disease in adulthood but rarely seen in pediatric patients. Calcification of a bicuspid aortic valve does not generally begin until the fourth decade of life, while sclerosis can be seen as early as the second decade in otherwise asymptomatic valves (31).
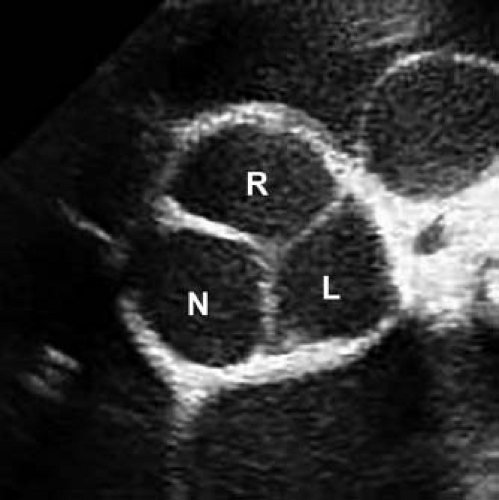 Figure 44.2 Echocardiographic parasternal short-axis view of a normal aortic valve in diastole. Right (R), left (L), and non- (N) coronary cusps are identified. |
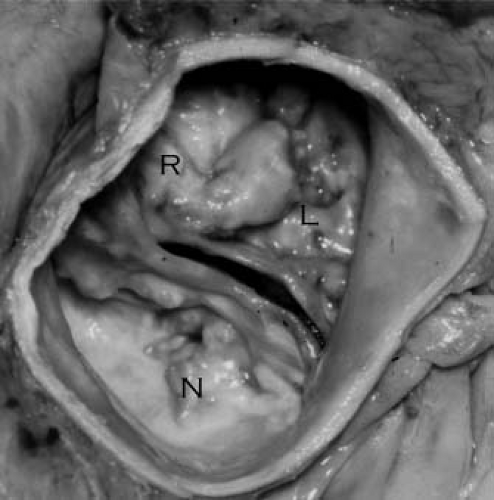 Figure 44.3 Pathologic specimen of a stenotic, bicuspid aortic valve. In the most common cases, there is fusion of the right (R) and left (L) coronary cusps with a thickened raphe at the site of the intracoronary commissure. N, non-coronary cusp. |
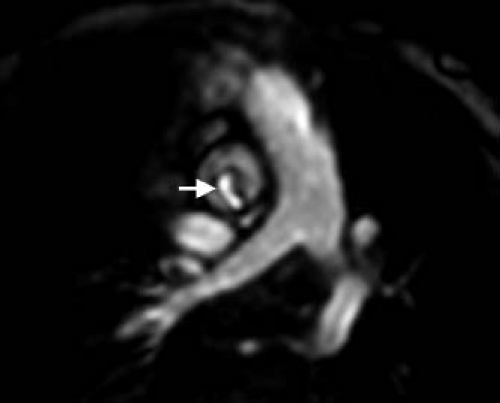 Figure 44.4 Cardiac MRI axial oblique image demonstrates a unicuspid, unicommissural aortic valve with a single, posteriorly oriented commissure (identified with an arrow). The patient is a 3-year-old girl with Turner syndrome status post coarctation repair. |
Dilation of the ascending aorta or aortic root is commonly seen in association with bicuspid aortic valves with or without significant stenosis (Fig. 44.6). At least mild dilation of the ascending aorta occurs in approximately 50% of pediatric and young adult patients with a bicuspid aortic valve, while at least mild dilation of the aortic root is found in 22% of this population (32). Moderate or severe dilation of the ascending aorta or aortic root is present in 16% and 5% of the patients, respectively. Significant stenosis does not appear to increase the risk of significant aortic dilation, and in fact, valves with moderate or greater aortic stenosis have been associated with less dilation of the aortic root than those with milder stenosis (32).
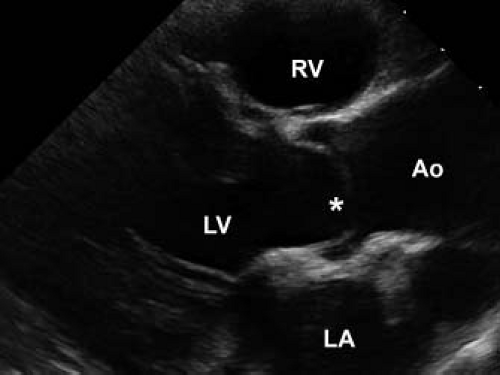 Figure 44.5 Echocardiographic parasternal long-axis view of a stenotic, bicuspid aortic valve. The doming tips of the valve cusps are indicated with an asterisk. The patient is a 11-year-old boy prior to balloon aortic valvuloplasty. RV, right ventricle; LV, left ventricle; Ao, aorta; LA, left atrium. |
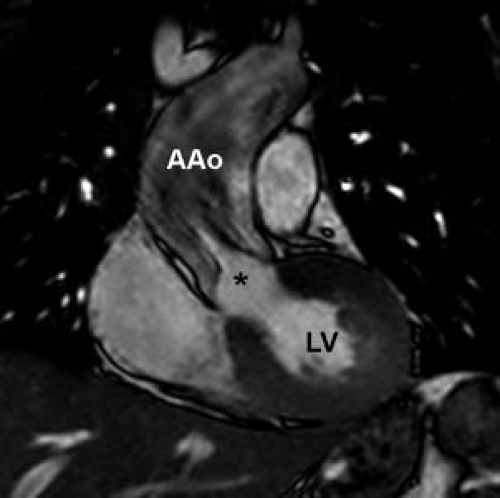 Figure 44.6 Cardiac MRI coronal oblique image demonstrates a doming aortic valve with dephasing jet at the leaflet tips (marked with an asterisk) due to turbulent flow. The ascending aorta is significantly dilated. The patient is a 22-year-old young man with a bicuspid aortic valve status post surgical valvotomy. AAo, ascending aorta; LV, left ventricle. |
Valvar aortic stenosis is associated with additional anomalies in up to 25% of published cases series, with coarctation of the aorta, patent ductus arteriosus, and ventricular septal defect being the most common (33). The association between coarctation of the aorta and bicuspid aortic valve has been well described, with a bicuspid valve being identified in 20% to 85% of cases of isolated coarctation (31). Concentric left ventricular hypertrophy is the physiologic response to left ventricular outflow tract obstruction, and is often observed in association with hemodynamically significant stenosis. In cases of severe neonatal aortic stenosis, obstruction to left ventricular outflow in utero often leads to the development of endocardial fibroelastosis, a diffuse thickening and scarring of the left ventricular endocardium (34,35).
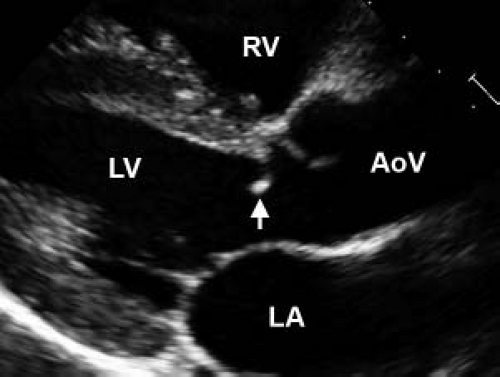 Figure 44.7 Echocardiographic parasternal long-axis view of a discrete subvalvar membrane. The thin membrane, indicated with an arrow, is located 3 mm below the aortic valve. The patient is a 14-year-old boy with isolated subvalvar aortic stenosis. RV, right ventricle; LV, left ventricle; Ao, Aorta; LA, left atrium. |
Subvalvar Aortic Stenosis
The pathology of subvalvar aortic stenosis is polymorphic in nature. The most common form, accounting for 70% to 80% of cases, is membranous subaortic stenosis, characterized by a thin, fibrous membrane just proximal to the aortic valve (Fig. 44.7, Video 44.4) (36). The membrane is generally very thin (1 to 2 mm), located within several millimeters of the aortic valve, and is often circumferential, including attachments to the anterior leaflet of the mitral valve (12,13,35). The second most common type of subaortic stenosis involves a fibromuscular ridge, thicker than the membrane and frequently located slightly more inferior to the aortic valve (Fig. 44.8, Video 44.5) (12,13,37). Both of these types of discrete subaortic stenosis are rarely present in infancy and are often considered acquired conditions. While their pathogenesis is incompletely understood, there is evidence that certain anatomic substrates create abnormal flow patterns and shear stresses within the left ventricular outflow tract that lead to endothelial damage, cellular proliferation, and collagen deposition resulting in the formation of the fibrous membrane or ridge. An abnormally small angle between the muscular and conal septae may also be a contributing factor to the necessary abnormalities in flow, and the presence of a perimembranous ventricular septal defect likely also predisposes to membrane formation (38,39,40,41,42). As the fibrous deposition becomes prominent enough to cause significant obstruction, the resultant left ventricular hypertrophy can cause the septum to further impinge on the left ventricular outflow tract leading to a self-sustaining cycle of obstruction and hypertrophy.
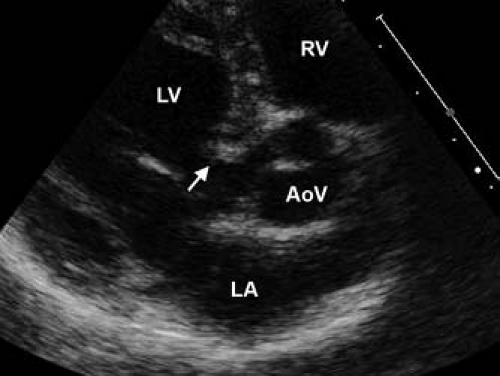 Figure 44.8 Echocardiographic parasternal long-axis view of a thick, fibromuscular ridge, indicated with an arrow, located 5 mm below the aortic valve. The patient is a 4-year-old boy with a bicuspid aortic valve, history of coarctation repair, and prior resection of a thin, subvalvar membrane. RV, right ventricle; LV, left ventricle; AoV, aortic valve; LA, left atrium. |
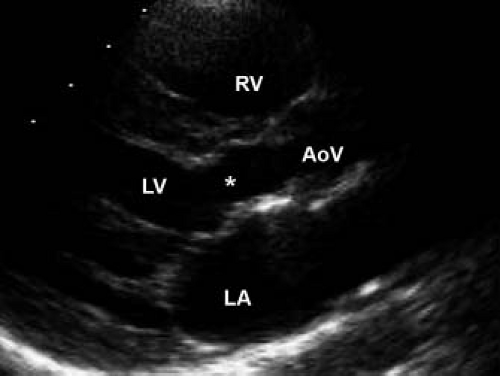 Figure 44.9 Echocardiographic parasternal long-axis view of tunnel-like narrowing of the left ventricular outflow tract. The narrowed region, indicated by an asterisk, extends for approximately 1.5 cm below the aortic valve annulus. The patient is a 13-year-old girl with a history of previous subaortic stenosis resection. RV, right ventricle; LV, left ventricle; AoV, aortic valve; LA, left atrium. |
On the most severe end of the subaortic stenosis disease spectrum is “tunnel-type” obstruction, characterized by muscular hypertrophy and narrowing of the left ventricular outflow tract that extends for several centimeters below the valve (Fig. 44.9, Video 44.6) (12,43). While tunnel type obstruction can present de novo (12,13), it is most commonly diagnosed in patients who have undergone prior congenital heart surgery (44). In the series reported by Jahangiri et al., patients with a diverse group of primary diagnoses developed complex or tunnel-like obstruction postoperatively. The most common primary diagnosis was double outlet right ventricle, while interrupted aortic arch, membranous subaortic stenosis, and Shone complex were also significant contributors. As with membranous subaortic stenosis, it is likely that abnormal flow patterns and shear stresses caused by even mild residual postoperative subaortic obstruction result in proliferation and hypertrophy leading to the tunnel-like obstruction. It may be that anatomic narrowing of the left ventricular outflow tract, as with posterior deviation of the infundibular ventricular septum in patients with interrupted aortic arch, may predispose to long segment stenosis as opposed to the more common discrete membranous stenosis. Decreased effective size of the ventricular septal defect, related either to shrinkage of intracardiac baffle material or fibrous deposition within a baffle, has also been proposed as an etiology of left ventricular outflow tract obstruction after repairs requiring construction of a left ventricle to aorta baffle (45).
Other etiologies of subaortic obstruction include anomalous insertions of mitral valve chordae or accessory atrioventricular valve tissue (often in the setting of a cleft mitral valve or complete atrioventricular septal defect), the left ventricular outflow tract “goose-neck deformity”
present in complete atrioventricular septal defects, posterior malalignment of the infundibular septum, and asymmetric septal hypertrophy as a subtype of hypertrophic cardiomyopathy (35,44,46,47).
present in complete atrioventricular septal defects, posterior malalignment of the infundibular septum, and asymmetric septal hypertrophy as a subtype of hypertrophic cardiomyopathy (35,44,46,47).
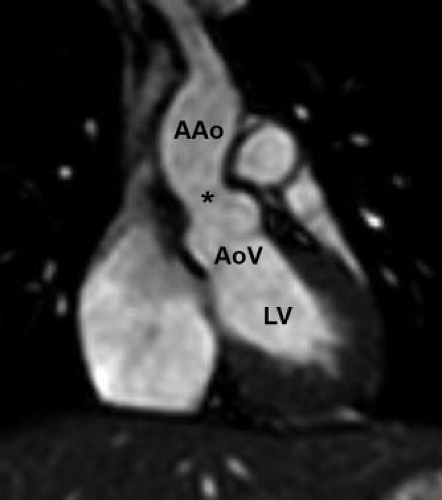 Figure 44.10 Cardiac MRI coronal oblique image demonstrates mild narrowing at the sinotubular junction (marked with an asterisk). The patient is a 16-year-old girl with Williams–Beuren syndrome. AAo, ascending aorta; AoV, aortic valve; LV, left ventricle. |
Another important associated finding is aortic insufficiency, which develops in up to 70% of patients with subaortic stenosis over time (36,48) (Videos 44.7 and 44.8, without and with color mapping). While in some cases aortic insufficiency is related to previous balloon or surgical intervention on the aortic valve (37), insufficiency may also develop in patients with isolated, discrete subvalvar stenosis (13,49). In these cases, the aortic regurgitation may be secondary to valve damage from long-standing exposure to the high-velocity jet caused by the subvalvar membrane. Consistent with this hypothesis, a higher peak echocardiographic Doppler gradient is the strongest independent risk factor for aortic insufficiency in patients with discrete subvalvar stenosis (37). While aortic valve morphology is most often normal, a bicuspid aortic valve is identified in 10% to 25% of patients (14,37). Additional cardiac malformations are frequent, most common being a ventricular septal defect, found in 10% to 48% of patients (50,51). Coarctation of the aorta is the next most common association, present in 6% to 20% of patients with subvalvar stenosis (13,37). Subvalvar aortic stenosis and coarctation of the aorta are sometimes accompanied by additional left-sided anomalies including a supravalvar mitral ring and a parachute mitral valve in a constellation initially described by Shone et al. (52).
Supravalvar Aortic Stenosis
Supravalvar aortic stenosis is a manifestation of ELN arteriopathy, a systemic process that may affect any artery in the body, but is generally most pronounced in the large arteries with high ELN content, most notably the aorta. ELN is a protein produced by multiple cell types including smooth muscle cells, endothelial cells, and various cells in the extracellular matrix (53). ELN is the primary component of the arterial extracellular matrix and combines with multiple other extracellular proteins to form the elastic fibers necessary for the characteristic distensibility of large arteries (54). These elastic fibers are organized in concentric rings, termed elastic lamellae, which surround the arterial lumen. Each elastic lamella is paired with a ring of smooth muscle, jointly referred to as a lamellar unit (55). ELN also plays a key role in arterial morphogenesis, regulating vascular smooth muscle proliferation and organization (56). ELN−/− mice display thickened aortae with near complete obliteration of the lumen due to accumulation of subendothelial smooth muscle cells (56). Pathological evaluation of tissue from humans with ELN arteriopathy demonstrates similar subendothelial smooth muscle accumulation as well as smooth muscle hypertrophy, fibrosis, collagen deposition, and disruption of elastic fibers at sites of stenosis (57,58,59,60). Importantly, the morphology of even nonstenotic arteries in patients with ELN arteriopathy is quite abnormal as well, with markedly thickened vessel walls due to increased numbers of lamellar units in the media layers (55,58). It is thought that the increased number of lamellar units in patients with ELN mutations is a physiologic means of maintaining constant wall stress in the setting of dysfunctional elastic fibers, but that eventually the number of units surpasses the potential blood supply leading to medial necrosis, increased fibrosis, and focal stenosis (55).
The classic description of supravalvar aortic stenosis involved discrete obstruction just distal to the sinuses of Valsalva (Fig. 44.10) (59,61). While this remains the most common presentation, the aortic phenotype of ELN arteriopathy is quite variable, ranging from mild stenosis at the sinotubular junction to severe, diffuse narrowing of the entire aorta (53,62). The most severe form of the disease, referred to as middle aortic syndrome, features narrowing of the thoracic and abdominal aorta as well as important branches including the mesenteric and renal arteries (63). Right ventricular outflow tract obstruction, another manifestation of ELN arteriopathy, is found in 40% to 80% of patients with supravalvar aortic stenosis (16,58). The most common type of right-sided obstruction in this setting is peripheral branch pulmonary artery stenosis, though more proximal branch pulmonary artery stenosis and supravalvar main pulmonary artery stenosis also occur (64,65,66). In addition to the pulmonary arteries, intracerebral arteries may also be affected by the diffuse arteriopathy with potential risk for spontaneous stroke (67,68).
The coronary arteries are an important potential site of disease in patients with ELN arteriopathy (69). As ELN-containing arteries, the coronaries do develop thickened walls with the potential for diffuse luminal narrowing (70,71), but there are several other mechanisms that also contribute to impaired coronary artery perfusion in patients with supravalvar aortic stenosis (58). The most common coronary anomaly in this population is ostial stenosis, which is present in up to 45% of patients with supravalvar aortic stenosis referred for surgery (16). More commonly affecting the left coronary artery than the right, ostial stenosis can be caused by adhesion of an aortic valve cusp to the supravalvar ridge immediately above the coronary orifice (71,72). In extreme cases, the ostium can be completely excluded from the aortic lumen by the valve tissue (73,74,75,76). Alternatively, ostial stenosis or even atresia can be due to pathologic thickening of the aortic or proximal coronary artery wall (71,77,78,79) that impinges on the orifice. While stenosis or obstruction is the most concerning coronary manifestation of supravalvar aortic stenosis, coronary artery dilation is also well described and is likely more common (66,70,80). Generally the dilation is not severe, but case reports of large coronary artery aneurysms in the setting of supravalvar stenosis have been documented (81) along with an increased potential risk for thrombosis.
Aortic valve pathology is common in cases of supravalvar aortic stenosis. A bicuspid aortic valve is identified in 8% to 25% of patients (16,17,82,83), while up to 50% of patients in some series have a stenotic or dysmorphic valve (58,76). Aortic insufficiency is present in approximately 10% to 30% of patients in larger series (76,83). The most common aortic valve abnormality in the setting
of supravalvar stenosis is restricted cusp excursion due to adhesions between valve leaflets and the supravalvar aortic ridge (69). As such, much of the aortic valve disease associated with supravalvar stenosis is likely not inherent to the valve itself, but rather secondary to degenerative changes related to this adhesion formation as well as altered biomechanics due to the reduced distensibility of the sinotubular junction (76). Other associated cardiac anomalies include coarctation of the aorta (10% to 12%), patent ductus arteriosus, patent foramen ovale or atrial septal defect, ventricular septal defect, and mitral valve abnormalities (16,17,82,84). As with the other types of aortic stenosis, concentric left ventricular hypertrophy often develops in the presence of significant obstruction.
of supravalvar stenosis is restricted cusp excursion due to adhesions between valve leaflets and the supravalvar aortic ridge (69). As such, much of the aortic valve disease associated with supravalvar stenosis is likely not inherent to the valve itself, but rather secondary to degenerative changes related to this adhesion formation as well as altered biomechanics due to the reduced distensibility of the sinotubular junction (76). Other associated cardiac anomalies include coarctation of the aorta (10% to 12%), patent ductus arteriosus, patent foramen ovale or atrial septal defect, ventricular septal defect, and mitral valve abnormalities (16,17,82,84). As with the other types of aortic stenosis, concentric left ventricular hypertrophy often develops in the presence of significant obstruction.
Molecular Genetics
Valvar Aortic Stenosis
A strong genetic contribution toward the development of bicuspid aortic valve, aortic stenosis, and other left-sided obstructive lesions has long been suspected based upon epidemiologic observations. Family clusters of bicuspid aortic valve (85,86,87,88), valvar aortic stenosis (89), coarctation of the aorta (90,91) and hypoplastic left heart syndrome (92,93) have been noted, and the presence of multiple types of left heart obstructive lesions in the same family has also been well described (94,95). The occurrence of multiple types of left-sided obstructive lesions within families has suggested that phenotypic heterogeneity along a broad spectrum of disease severity may result from common genetic causes. Multiple gene interactions, variable expressivity, incomplete penetrance, and the influence of environmental and epigenetic factors all likely contribute to the diverse pedigrees reported in the literature (96).
The suggestion from these initial case series that disease of the aortic valve and left ventricular outflow tract is both highly heritable and phenotypically diverse has subsequently been confirmed by larger genetic studies. An early analysis of familial aggregation of congenital heart disease demonstrated that first-degree relatives of probands with left ventricular outflow tract obstruction (aortic coarctation or hypoplastic left heart syndrome) were more likely to have congenital heart disease than relatives of probands with d-transposition of the great arteries (19.3% and 9.4% vs. 2.7% of first-degree relatives affected, respectively) (97). Statistical modeling based on pedigrees of patients with bicuspid aortic valve and hypoplastic left heart syndrome has suggested that each condition is highly heritable, with segregation patterns of both anomalies estimated to be almost entirely due to genetic factors (heritability 0.89 and 0.99, respectively) (98,99). Congenital heart malformations were identified in 18% of first-degree relatives of probands with hypoplastic left heart syndrome, and the great majority (78%) of the heart disease consisted of left heart obstructive lesions. McBride et al. (100) confirmed a similarly high estimate of heritability (0.71 to 0.90) in an analysis of patients with assorted left ventricular outflow tract obstructive lesions (aortic stenosis, coarctation of the aorta, and hypoplastic left heart). Left-sided obstructive lesions were identified in 7.3% of first-degree relatives of probands in this study, which was likely an underestimate as asymptomatic aortic valve disease may have been missed in the 52 relatives (12.3%) who did not have echocardiography data available. The relative risk for left ventricular outflow tract obstruction for first-degree relatives of probands in this analysis was 36.9. Asymptomatic bicuspid aortic valves were identified in 4.7% of first-degree relatives of patients with nonsyndromic left ventricular outflow tract obstruction in another study, with the prevalence of asymptomatic bicuspid valve increasing to 16.7% in families with more than one affected individual (101).
While the highly heritable nature of congenital left heart obstruction has been clearly established, the underlying molecular genetic mechanisms have not been fully elucidated. Several syndromes are known to involve aortic valve anomalies, most notably Turner syndrome, a chromosomal disorder characterized by complete or partial absence of one X chromosome in a female. A bicuspid aortic valve is present in 30% of patients with Turner syndrome, and clinically significant aortic disease (coarctation, aortic stenosis) is found in approximately 10% of affected newborns (102). While the causative gene has not been identified, analysis of the phenotypes of patients with partial deletions of an X chromosome suggest that the relevant genetic material is on the chromosome’s short arm, Xp (102). Another chromosomal disorder linked to aortic valve disease is 11q terminal deletion disorder, also known as Jacobsen syndrome. More than half of patients with Jacobsen syndrome have congenital heart disease, with approximately 18% of patients having left-sided obstructive lesions (103). The critical region on 11q for left ventricular outflow tract development has not yet been identified, and the gene deletion(s) responsible for left heart obstruction in this setting are not yet known.
Linkage analyses performed on families with inherited bicuspid aortic valve have identified multiple associated genes including TGFBR1, TGFBR2, ACTA2, KCNJ2, FLNB, KMT2D, and KDM6A (96). Each of these genes is related to a syndrome known to include bicuspid aortic valve as part of its phenotype (Table 44.1), but the molecular mechanisms underlying the valve disease in these cases have not been determined. To date, the only gene to be linked to isolated aortic valve disease is NOTCH1 (104). The Notch signaling pathway, one of the primary pathways downstream of VEGF, a crucial regulator of angiogenesis and endothelial cell differentiation, is known to play a key role in cardiovascular development (105). In the first two families in which NOTCH1 mutations were identified, affected members had a relatively high incidence of asymptomatic bicuspid aortic valve, aortic stenosis, and hypoplastic left heart syndrome (104). Interestingly, the pedigrees also included adults with calcific stenosis affecting tricuspid aortic valves, suggesting a role for NOTCH1 in both valve development and subsequent valve maintenance. The mechanism by which NOTCH1 mutations impact aortic valve morphogenesis has not been fully characterized, but there is in vitro evidence that NOTCH1 mutant alleles from patients with left ventricular outflow tract malformations are associated with impaired EMT, a process necessary for endothelial cushion formation and semilunar valve formation (discussed further in Embryology and Pathology section) (106). In murine models, defective NOTCH signaling has been shown to inhibit EMT and disrupt the migration of neural crest cells necessary for outflow tract development (107). Murine models have also revealed a role for NOTCH signaling in aortic valve homeostasis with NOTCH1 acting to prevent premature calcification via inhibition of BMP2 (108). This finding was consistent with the observation, in the original description of the role of NOTCH1 in human aortic valve disease, that NOTCH1 repressed RUNX2, which is another key mediator of calcium deposition within the valve tissue (104).
Another link between aortic valve morphogenesis and the subsequent development of calcific aortic valve disease involves an interaction between NOTCH1 and endothelium-derived nitric oxide synthase (eNOS). Supplementation with nitric oxide has been shown to prevent calcification of aortic valve interstitial cells in vitro (109), while nitric oxide deficiency is associated with bicuspid valve formation in mice (109). Recently, nitric oxide has been shown to modulate the nuclear localization of the NOTCH1 intracellular domain in cultured aortic valve interstitial cells, as well as regulate the expression of HEY1, one of NOTCH1 downstream targets. In addition, knocking out both eNOS and NOTCH1 in a murine model results in highly penetrant aortic valve disease. This suggests interaction between the two pathways in aortic valve morphogenesis (110).
One intriguing potential connection between nitric oxide and aortic valve development involves the observation that mechanical factors, such as decreased flow in developing hearts, can contribute to abnormalities of the aortic valve and left ventricular outflow tract. Premature closure of the patent foramen ovale in the first
trimester, with resultant decreased flow through the left heart, has been linked to aortic atresia and hypoplastic left heart syndrome in humans (111), while disrupting flow in the left heart in chick embryos can also induce the development of hypoplastic left heart syndrome (112). It has been demonstrated that shear stress increases expression of eNOS (113,114), and it may be that alteration in nitric oxide synthesis by endothelial cells in response to changes in flow dynamics could explain some of the influence that hemodynamics and blood flow appear to have on development of the left ventricular outflow tract. Similarly, the unique geometry of a bicuspid aortic valve has also been shown to result in alterations in shear stress compared to a tricuspid aortic valve (115), and this is thought to contribute to the early development of calcific aortic stenosis in bicuspid valves. Nitric oxide metabolism may play a role in this process as well.
trimester, with resultant decreased flow through the left heart, has been linked to aortic atresia and hypoplastic left heart syndrome in humans (111), while disrupting flow in the left heart in chick embryos can also induce the development of hypoplastic left heart syndrome (112). It has been demonstrated that shear stress increases expression of eNOS (113,114), and it may be that alteration in nitric oxide synthesis by endothelial cells in response to changes in flow dynamics could explain some of the influence that hemodynamics and blood flow appear to have on development of the left ventricular outflow tract. Similarly, the unique geometry of a bicuspid aortic valve has also been shown to result in alterations in shear stress compared to a tricuspid aortic valve (115), and this is thought to contribute to the early development of calcific aortic stenosis in bicuspid valves. Nitric oxide metabolism may play a role in this process as well.
TABLE 44.1 Genetic Causes of Bicuspid Aortic Valve and Aortic Stenosis | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
Subvalvar Stenosis
Though often considered an acquired condition, several familial clusters of discrete subvalvar aortic stenosis have been reported, suggesting a genetic basis of disease in at least some cases. While no causative genes have been identified, inheritance patterns suggest autosomal recessive transmission may be involved in some instances. Of the ten families with inherited discrete subvalvar stenosis reported in the literature, four consisted of unaffected consanguineous parents with multiple affected children, consistent with autosomal recessive inheritance (116,117,118). The remainder of the reported pedigrees were suggestive of autosomal dominant inheritance, but limited numbers of affected individuals within the pedigrees make interpretation difficult (118). While the existence of these family clusters indicates that genetic factors play a role in the development of subvalvar stenosis in some instances, the rarity of these familial cases suggests that the heritability of subvalvar stenosis is in general far less than either valvar or supravalvar aortic disease.
One additional piece of evidence suggesting some genetic influence in the development of subvalvar stenosis is the overrepresentation of bicuspid aortic valve (up to 25%) among patients with subvalvar obstruction (37). As discussed previously, bicuspid aortic valve is a highly heritable condition, with an overall prevalence of approximately 1% in the general population. The relatively high prevalence of bicuspid aortic valve among patients with subvalvar stenosis strongly suggests a genetic component in susceptibility to the disease. Given the hypothesized role for abnormal flow and shear stress in the formation of subvalvar membranes, however, it may simply be that any abnormality of the left ventricular outflow tract that provides a substrate for nonlaminar flow may result in membrane formation.
Supravalvar Stenosis
The molecular genetics of supravalvar aortic stenosis have been fairly well described. As discussed above, aortic disease is the most prominent manifestation of a systemic arteriopathy. Supravalvar aortic stenosis has been traditionally associated with Williams–Beuren syndrome (61,74,119), a syndrome in which the arteriopathy is accompanied by cognitive disability, distinctive “elfin facies,” hypocalcemia, small stature, and an ebullient personality (69). Identical vascular pathology is also seen in the absence of the other findings of Williams–Beuren syndrome, however, both in inherited and sporadic forms (120,121). Careful genetic studies have now linked both syndromic and nonsyndromic supravalvar aortic stenosis to haploinsufficiency of the ELN gene.
ELN, located on chromosome 7q11.23, was first identified as a candidate gene by linkage studies performed on two families with inherited supravalvar aortic stenosis that mapped the disease-causing gene to the long arm of chromosome 7 (122). Soon afterward, a balanced translocation disrupting the ELN gene was identified in a family with autosomal dominant supravalvar stenosis, further strengthening the association between the arteriopathy and ELN deficiency (123). Subsequently, the supravalvar aortic stenosis phenotype has been produced in heterozygous ELN gene knockout mice (55), while multiple additional ELN mutations have been identified in patients with both sporadic and familial supravalvar stenosis (15,124). Familial supravalvar aortic stenosis is generally inherited as an autosomal dominant trait. Echocardiographic
screening of asymptomatic family members of affected individuals suggests that the disease is highly penetrant with variable expressivity accounting for diverse phenotypes (125).
screening of asymptomatic family members of affected individuals suggests that the disease is highly penetrant with variable expressivity accounting for diverse phenotypes (125).
Confirming the similarities in vascular phenotype between syndromic and nonsyndromic supravalvar aortic stenosis, Williams–Beuren syndrome has also been linked to the ELN gene. Williams–Beuren syndrome is now known to be caused by a microdeletion of approximately 1.55 megabases at chromosome 7q11.23, with 26 to 28 genes including ELN generally being deleted (53,62,126,127). While the non-ELN deleted genes are responsible for the noncardiac manifestations of the syndrome, the molecular consequence of the ELN-deficient arteriopathy is identical in syndromic and nonsyndromic cases of supravalvar aortic stenosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


