Chapter 35
Antithrombotic Therapy
Iqbal H. Jaffer, Jeffrey I. Weitz
Arterial and venous thromboses are major causes of morbidity and mortality. Arterial thrombosis is the most common cause of acute myocardial infarction (MI), ischemic stroke, and limb gangrene, whereas deep vein thrombosis can lead to postthrombotic syndrome and pulmonary embolism, which can be fatal. Most arterial thrombi are superimposed on disrupted atherosclerotic plaque, which exposes thrombogenic material in the plaque core to the blood. This material then triggers platelet aggregation and fibrin formation, which results in the generation of a platelet-rich thrombus that can temporarily or permanently occlude blood flow.1 In contrast to arterial thrombi, venous thrombi rarely form at sites of obvious vascular disruption. Although they can develop after surgical trauma to veins, or secondary to indwelling venous catheters, venous thrombi usually originate in the valve cusps of the deep veins of the calf or in the muscular sinuses,2 where they are triggered by stasis. Sluggish blood flow in these veins reduces the oxygen supply to the avascular valve cusps.3 Endothelial cells lining these valve cusps become activated and express adhesion molecules on their surface. Tissue factor–bearing leukocytes and microparticles adhere to these activated cells and induce coagulation.4 In addition, webs of DNA released from activated neutrophils, so-called neutrophil extracellular traps, also contribute to thrombosis by providing a scaffold that binds platelets and promotes their activation and aggregation.5 Local thrombus formation is exacerbated by reduced clearance of activated clotting factors as a result of impaired blood flow. If the calf vein thrombi extend into more proximal veins of the leg, thrombus fragments can dislodge, travel to the lungs, and produce a pulmonary embolism.6
Arterial and venous thrombi are composed of platelets and fibrin, but the proportions differ. Arterial thrombi are rich in platelets because of the high shear in the injured arteries. In contrast, venous thrombi, which form under low shear conditions, contain relatively few platelets and are predominantly composed of fibrin and trapped red cells.7 Because of the predominance of platelets, arterial thrombi appear white, whereas venous thrombi are red in color reflecting the trapped red cells.
Antithrombotic drugs are used for prevention and treatment of thrombosis. Targeting the components of thrombi, these agents include (a) antiplatelet drugs, which inhibit platelets; (b) anticoagulants, which attenuate coagulation; and (c) thrombolytic agents, which induce fibrin degradation (Fig. 35-1). With the predominance of platelets in arterial thrombi, strategies to inhibit or treat arterial thrombosis focus mainly on antiplatelet agents, although in the acute setting, they often include anticoagulants and thrombolytic agents. Anticoagulants are the mainstay of prevention and treatment of venous thromboembolism because fibrin is the predominant component of venous thrombi. Antiplatelet drugs are less effective than anticoagulants in this setting because of the limited platelet content of venous thrombi. Thrombolytic therapy is used in selected patients with venous thromboembolism. For example, patients with massive or submassive pulmonary embolism can benefit from systemic or catheter-directed thrombolysis. Catheter-directed thrombolysis also can be used as an adjunct to anticoagulants for treatment of certain patients with extensive iliofemoral vein thrombosis.8

This chapter focuses on antiplatelet and anticoagulant drugs. In addition to agents in current use, new drugs that are in advanced stages of development also will be discussed. Thrombolytic therapy is discussed in Chapter 36.
Antiplatelet Drugs
Role of Platelets in Arterial Thrombosis
In healthy vasculature, circulating platelets are maintained in an inactive state by nitric oxide and prostacyclin released by endothelial cells lining the blood vessels. In addition, endothelial cells also express ADPase on their surface, which degrades adenosine diphosphate (ADP) released from activated platelets. When the vessel wall is damaged, release of these substances is impaired and subendothelial matrix is exposed.9 Platelets adhere to exposed collagen, von Willebrand factor (vWF), and fibronectin via α2β1, glycoprotein Ib/IX (GP-Ib/IX), and α5β1, respectively—receptors constitutively expressed on the platelet surface. Adherent platelets undergo a change in shape, secrete ADP from their dense granules, and synthesize and release thromboxane A2 (TXA2). Released ADP and TXA2, which are platelet agonists, activate ambient platelets and recruit them to the site of vascular injury (Fig. 35-2).
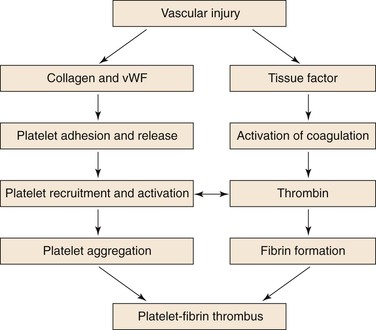
Figure 35-2 Coordinated role of platelets and the coagulation system in thrombogenesis. Vascular injury simultaneously triggers platelet activation and aggregation, as well as activation of the coagulation system. Platelet activation is initiated by exposure of subendothelial collagen and von Willebrand factor (vWF) onto which platelets adhere. Adherent platelets become activated and release ADP and TXA2, platelet agonists that activate ambient platelets and recruit them to the site of injury. When platelets are activated, glycoprotein IIb/IIIa on their surface undergoes a conformational change that enables it to ligate fibrinogen and mediate platelet aggregation. Coagulation is triggered by tissue factor exposed at the site of injury. Tissue factor triggers thrombin generation. As a potent platelet agonist, thrombin amplifies platelet recruitment to the site of injury. Thrombin also converts fibrinogen to fibrin; the fibrin strands then weave the platelet aggregates together to form a platelet/fibrin thrombus.
Disruption of the vessel wall also exposes tissue factor–expressing cells to the blood. Tissue factor initiates coagulation. Activated platelets potentiate coagulation by binding clotting factors and supporting the assembly of activation complexes that enhance thrombin generation. In addition to converting fibrinogen to fibrin, thrombin also serves as a potent platelet agonist and recruits more platelets to the site of vascular injury. Greater detail on the role of activated platelets and coagulation is presented in Chapter 34.
When platelets are activated, GP-IIb/IIIa, the most abundant receptor on the platelet surface, undergoes a conformational change that enables it to ligate fibrinogen. Divalent fibrinogen molecules bridge adjacent platelets together to form platelet aggregates. Fibrin strands, generated through the action of thrombin, then weave these aggregates together to form a platelet/fibrin mesh.
Antiplatelet drugs target various steps in this process. The commonly used drugs include aspirin, the thienopyridines (ticlopidine, clopidogrel, and prasugrel), ticagrelor, and GP-IIb/IIIa antagonists. Each is briefly described.
Aspirin
The most widely used antiplatelet agent worldwide is aspirin. As a cheap and effective antiplatelet drug, aspirin serves as the foundation of most antiplatelet strategies.
Mechanism of Action
Aspirin produces its antithrombotic effect by irreversibly acetylating and inhibiting platelet cyclooxygenase (COX)-1 (Fig. 35-3), a critical enzyme in the biosynthesis of TXA2. At high doses (about 1 g per day), aspirin also inhibits COX-2, an inducible COX isoform found in endothelial cells and inflammatory cells.9 In endothelial cells, COX-2 initiates the synthesis of prostacyclin, a potent vasodilator and inhibitor of platelet aggregation.
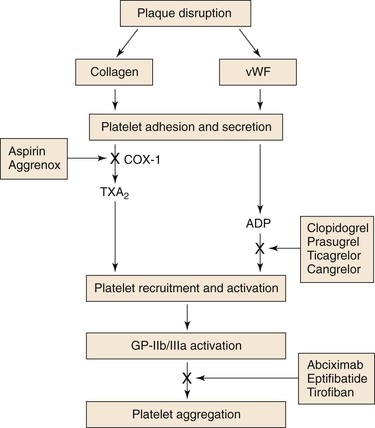
Figure 35-3 Site of action of antiplatelet drugs. Aspirin inhibits TXA2 synthesis by irreversibly acetylating cyclooxygenase-1 (COX-1). Reduced TXA2 release attenuates platelet activation and recruitment to the site of vascular injury. Ticlopidine, clopidogrel, and prasugrel irreversibly block P2Y12, a key ADP receptor on the platelet surface, whereas ticagrelor acts as a reversible inhibitor of P2Y12. Therefore these agents also attenuate platelet recruitment. Abciximab, eptifibatide, and tirofiban inhibit the final common pathway of platelet aggregation by blocking fibrinogen binding to activated glycoprotein (GP) IIb/IIIa.
Indications
Aspirin is widely used for secondary prevention in patients with established coronary, cerebrovascular, or peripheral arterial disease. In such patients, aspirin produces a 25% reduction in the risk for cardiovascular death, MI, or stroke.10 The use of aspirin for primary prevention is more controversial. Recent studies have questioned whether the benefits of daily aspirin for primary cardiac protection outweigh its associated risks for gastrointestinal and intracerebral hemorrhage.11,12 Consequently, aspirin is no longer recommended for primary cardiac prevention unless the baseline cardiovascular risk is higher than 10% at 10 years.
Dosages
Aspirin is usually administered at doses of 75 to 325 mg once daily. There is no evidence that higher-dose aspirin is more effective than lower aspirin doses, and some analyses suggest reduced efficacy with higher doses.13 Because the side effects of aspirin are dose related, daily aspirin doses of 75 to 150 mg are recommended for most indications. When rapid platelet inhibition is required, an initial aspirin dose of at least 160 mg should be given.13
Side Effects
Most common side effects are gastrointestinal and range from dyspepsia to erosive gastritis or peptic ulcers with bleeding and perforation.13 These side effects are dose related, and use of enteric-coated or buffered aspirin in place of plain aspirin does not eliminate them. The overall risk for major bleeding with aspirin ranges from 1% to 3% per year,13 and the risk increases when aspirin is given in conjunction with other antiplatelet drugs, such as clopidogrel, or with anticoagulants, such as warfarin. When dual therapy is used, low-dose aspirin should be given (75 to 100 mg daily). Eradication of Helicobacter pylori infection and administration of proton pump inhibitors reduce the risk for aspirin-induced gastrointestinal bleeding in patients with peptic ulcer disease.
Aspirin should not be administered to patients with a history of aspirin allergy characterized by bronchospasm. This problem occurs in about 0.3% of the general population, but it is more common in those with chronic urticaria or asthma, particularly in patients with coexisting nasal polyps or chronic rhinitis.14 Hepatic and renal toxicity are observed with aspirin overdose.
Aspirin Resistance
The term aspirin resistance has been used to describe both clinical and laboratory phenomena.15 Clinical aspirin resistance is defined as the failure of aspirin to protect patients from ischemic vascular events. This is not a helpful definition, because it is made after the event occurs. Furthermore, it is not realistic to expect aspirin, which only blocks TXA2-induced platelet activation, to prevent all vascular events.
Aspirin resistance has also been described biochemically as failure of the drug to produce its expected inhibitory effects on platelet TXA2 synthesis16 or arachidonic acid-induced platelet aggregation.17 However, the tests used for diagnosis of biochemical aspirin resistance have not been well standardized,15 and there is no definitive evidence that the tests identify patients at risk for recurrent vascular events or that resistance can be reversed by giving higher doses of aspirin or by adding other antiplatelet drugs.15 Until this information is available, testing for aspirin resistance remains a research tool.
Perioperative Aspirin Management
Aspirin does not need to be stopped before diagnostic tests or surgical procedures associated with a low risk for bleeding. These include dental procedures, such as tooth extraction or root canal surgery, dermatologic procedures, such as excision of basal or squamous cell cancers or malignant or premalignant nevi, or ophthalmologic procedures, such as cataract surgery. Although continuation of aspirin during major surgery increases the risk for bleeding, discontinuation of aspirin is associated with about a threefold increased risk for cardiovascular events in patients undergoing noncardiac surgery.18,19 Therefore, benefit-to-risk assessments must weigh the benefit of continued aspirin for reduction of cardiac events against the risk for increased bleeding at the time of surgery. In general, therefore, aspirin is discontinued 7 to 10 days before noncardiac surgery or diagnostic or therapeutic interventions associated with a moderate or high risk for bleeding, provided that the risk for cardiovascular events off aspirin is low. If the risk for cardiovascular events is high, aspirin can be continued because even though aspirin increases the risk for bleeding, the bleeding rarely requires medical intervention. Patients undergoing coronary artery bypass grafting (CABG) should continue aspirin up to the time of surgery because aspirin use within 5 days of CABG lowers the risk for postoperative death without increasing blood transfusion requirements or increasing the risk for reoperation.20 If aspirin is discontinued before surgery, it can usually be safely restarted 24 hours after surgery, provided that hemostasis has been secured.
P2Y12 Antagonists
Drugs that target P2Y12, a key ADP receptor on platelets, include the thienopyridines—ticlopidine, clopidogrel, and prasugrel—and ticagrelor. Of the thienopyridines, clopidogrel is preferred over ticlopidine because of enhanced safety and tolerability. Because ticlopidine is rarely used, it will not be discussed.
Mechanism of Action
The thienopyridines are structurally related drugs that selectively inhibit ADP-induced platelet aggregation by irreversibly blocking P2Y12 (see Fig. 35-3). Clopidogrel and prasugrel are both prodrugs that must be metabolized by the hepatic cytochrome P450 (CYP) enzyme system to acquire activity. Metabolic activation of clopidogrel is inefficient, because only 15% of absorbed drug undergoes activation through two oxidative steps. In addition, varying absorption, and loss-of-function polymorphisms in the enzymes responsible for clopidogrel activation or drug-drug competition for these enzymes may delay and decrease the antiplatelet effects of clopidogrel.21 Although prasugrel, a third-generation thienopyridine, also requires metabolic activation, the process is more efficient and predictable than the activation of clopidogrel. Consequently, prasugrel has a faster onset of action than clopidogrel and produces greater inhibition of ADP-induced platelet aggregation.22
Ticagrelor has a mechanism of action distinct from that of the thienopyridines. In contrast to clopidogrel and prasugrel, ticagrelor does not require metabolic activation and serves as a reversible inhibitor of P2Y12. Without a requirement for activation, ticagrelor has a more rapid onset of action than clopidogrel and a more rapid offset of action than either clopidogrel or prasugrel.
Indications
When compared with aspirin in patients with recent ischemic stroke, MI, or peripheral arterial disease, clopidogrel reduced the risk for cardiovascular death, MI, and stroke by 8.7%.23 Therefore clopidogrel is marginally more effective than aspirin, but it also is more expensive. In some patients, clopidogrel and aspirin are combined to capitalize on their capacity to block complementary pathways of platelet activation. For example, the combination of aspirin plus clopidogrel is recommended for at least 4 weeks after implantation of a bare metal stent in a coronary artery and for 1 year in those with a drug-eluting stent.24 Concerns about late in-stent thrombosis with drug-eluting stents25,26 have led some experts to recommend long-term use of clopidogrel plus aspirin for this indication, provided that the risk for bleeding is low.27
The combination of clopidogrel and aspirin also is effective in medically managed patients with unstable angina. Thus in 12,562 such patients, the risk for cardiovascular death, MI, or stroke was 9.3% in those randomized to the combination of clopidogrel and aspirin and 11.4% in those given aspirin alone. This 20% relative risk reduction with combination therapy was highly statistically significant.28 However, combining clopidogrel with aspirin increases the risk for major bleeding to about 2% per year. This bleeding risk persists even if the daily dose of aspirin is 100 mg or less.29 Therefore the combination of clopidogrel and aspirin should only be used when there is a clear benefit. For example, this combination has not proven to be superior to clopidogrel alone in patients with acute ischemic stroke,30 or to aspirin alone for primary prevention in those at risk for cardiovascular events.31
Prasugrel was compared with clopidogrel in 13,608 patients with moderate-risk to high-risk acute coronary syndrome scheduled for PCI.32 All patients received aspirin. The primary efficacy endpoint, a composite of cardiovascular death, nonfatal MI, or stroke, occurred in 12.1% of patients given clopidogrel and in 9.9% of those randomized to prasugrel. This 19% relative risk reduction and 2.2% absolute reduction was mainly due to a reduction in nonfatal MI. The rate of major bleeding was higher with prasugrel than with clopidogrel (1.4% and 0.9%, respectively), as was the rate of fatal bleeding (0.4% and 0.1%, respectively). When compared with clopidogrel in 7243 acute coronary syndrome patients who were managed medically and did not undergo cardiac revascularization, prasugrel did not significantly reduce the risk for cardiovascular death, MI, or stroke; rates of major bleeding were similar with prasugrel and clopidogrel.33 Based on the results of this study, prasugrel should not be used in place of clopidogrel in acute coronary syndrome patients who are medically managed, but is an option in higher-risk patients undergoing PCI.
Ticagrelor was compared with clopidogrel in 18,624 patients with acute coronary syndrome all of whom also received aspirin.34 At 12 months, ticagrelor produced a 16% reduction in the primary efficacy outcome; a composite of vascular death, MI, or stroke (from 11.7% to 9.8%). Ticagrelor also was associated with a 21% reduction in the rate of cardiovascular death (from 5.1% to 4.0%) and all-cause mortality (from 5.9% to 4.5%). Although ticagrelor did not increase the overall rate of major bleeding as defined in the trial, it was associated with a 19% increase in non-CABG surgery related major bleeding (from 3.8% to 4.5%). Based on these results, current guidelines recommend ticagrelor as a potential alternative to clopidogrel in acute coronary syndrome patients regardless of whether they are managed medically or with PCI.
Dosing
Clopidogrel is given once daily at a dose of 75 mg.24 Because its onset of action is delayed for several days, loading doses of clopidogrel are given when rapid ADP receptor blockade is desired. For example, patients undergoing coronary stenting are often given a loading dose of 300 to 600 mg, which produces inhibition of ADP-induced platelet aggregation in about 4 to 6 hours.24
Prasugrel is initiated with a loading dose of 60 mg followed by a maintenance dose of 10 mg once daily. A dose of 5 mg once daily is used for those over the age of 75 years or those with a body weight less than 60 kg. Prasugrel should be avoided in patients with a history of stroke or transient ischemic attack.
In contrast to the thienopyridines, which produce irreversible inhibition of P2Y12, ticagrelor inhibits the receptor in a reversible fashion. With a half-life of only about 8 hours, ticagrelor must be administered twice daily. Ticagrelor is initiated with a loading dose of 180 mg followed by a maintenance dose of 90 mg twice daily.
Side Effects
The major side effect of clopidogrel, prasugrel, and ticagrelor is bleeding. Gastrointestinal and hematologic side effects are rare with the thienopyridines.35 Side effects of ticagrelor include dyspnea, which is usually mild and dose-related, asymptomatic bradycardia with ventricular pauses, and a modest increase in the levels of uric acid.36,37
Clopidogrel Resistance
The capacity of clopidogrel to inhibit ADP-induced platelet aggregation varies among patients.38 This variability reflects, at least in part, genetic polymorphisms in the CYP isoenzymes involved in its metabolic activation. Most important of these isoenzymes is CYP2C19. Clopidogrel-treated patients with the loss-of-function CYP2C19*2 allele exhibit reduced platelet inhibition compared with those with the wild-type CYP2C19*1 allele; they also experience a higher rate of cardiovascular events.34,39,40 This is important because estimates suggest that up to 25% of whites, 30% of blacks, and 50% of Asians carry the loss-of-function allele, which would render them resistant to clopidogrel. Even patients with the reduced-function CYP2C19*3, *4, or *5 alleles may derive less benefit from clopidogrel than those with the full-function CYP2C19*1 allele. Patients with polymorphisms in ABCB1 may exhibit reduced clopidogrel absorption, and polymorphisms in CYP3A4 can also reduce the metabolic activation of clopidogrel. Polymorphisms in both of these enzymes have been linked to adverse clinical outcomes.41 In contrast to their effect on the metabolic activation of clopidogrel, CYP2C19 and CYP3A4 polymorphisms appear to be less important determinants of the activation of prasugrel.42 Thus in the study that compared prasugrel with clopidogrel in patients with acute coronary syndrome, an association was found between loss-of-function alleles and an increased rate of cardiovascular events with clopidogrel, but not with prasugrel.43 Similar results were observed when ticagrelor was compared with clopidogrel.44 Because of these observations, some guidelines recommend ticagrelor or prasugrel over clopidogrel for patients with acute coronary syndrome.
Concomitant administration of clopidogrel with proton-pump inhibitors, which are inhibitors of CYP2C19, may also produce a small reduction in the inhibitory effects of clopidogrel on ADP-induced platelet aggregation. Although this interaction does not appear to be clinically important,45 the U.S. Federal Drug Administration discourages the use of proton pump inhibitors, particularly omeprazole, in clopidogrel-treated patients.
The observation that genetic polymorphisms affecting clopidogrel metabolism influence clinical outcomes prompted investigation into the possibility that pharmacogenetic profiling and/or point-of-care platelet function testing could be used to identify clopidogrel-resistant patients and target them for more intensive antiplatelet therapy. Although up to 30% of clopidogrel-treated patients have evidence of reduced responsiveness to the drug, randomized clinical trials have failed to show that more intensive antiplatelet therapy improves outcome in such patients.46,47 Consequently, there is no indication for clopidogrel resistance testing at this time.
Perioperative Management
The risk for bleeding during surgery is higher with the P2Y12 inhibitors than with aspirin. Consequently, patients who are receiving clopidogrel or ticagrelor should discontinue the medication 7 to 10 days (minimum of 5 days) before surgery. Because of its shorter offset of action, ticagrelor needs to be held for only 5 days before surgery, although even 3 days is likely to be sufficient. Whenever possible, elective surgery in patients receiving dual antiplatelet therapy for coronary stent implantation should be deferred for at least 6 weeks after bare metal stent implantation and 12 months after drug-eluting stent implantation. If such patients require urgent surgery within 6 weeks of stent placement, dual antiplatelet therapy should be continued during the perioperative period. Clopidogrel is safer than prasugrel or ticagrelor in this setting.
Patients receiving dual antiplatelet therapy who are at risk for cardiovascular events should continue aspirin up to the time of surgery, but they should discontinue their thienopyridine 7 to 10 days before surgery. Ticagrelor can be stopped 5 days before surgery.
Dipyridamole
Dipyridamole is a relatively weak antiplatelet agent on its own,24 but an extended-release formulation of dipyridamole combined with low-dose aspirin, a preparation known as Aggrenox, is used for prevention of stroke in patients with transient ischemic attacks.48
Mechanism of Action
By inhibiting phosphodiesterase, dipyridamole blocks the breakdown of cAMP. Increased levels of cAMP reduce intracellular calcium and inhibit platelet activation. Dipyridamole also blocks the uptake of adenosine by platelets and other cells. With more extracellular adenosine, local cAMP levels are increased further because the platelet adenosine A2 receptor is coupled to adenylate cyclase.
Dosing
Aggrenox is given twice daily. Each capsule contains 200 mg of extended-release dipyridamole and 25 mg of aspirin.48
Side Effects
Because dipyridamole has vasodilatory effects, it must be used with caution in patients with coronary artery disease. Gastrointestinal complaints, headache, facial flushing, dizziness, and hypotension also can occur. These symptoms often subside with continued use of the drug.48
Indications
Dipyridamole plus aspirin was compared with aspirin or dipyridamole alone, or with placebo, in patients with an ischemic stroke or transient ischemic attack. The combination reduced the risk for stroke by 22.1% compared with aspirin and by 24.4% compared with dipyridamole.49 A second trial compared dipyridamole plus aspirin with aspirin alone for secondary prevention in patients with ischemic stroke. Vascular death, stroke, or MI occurred in 13% of patients given combination therapy and in 16% of those treated with aspirin alone.50 Based on these data, Aggrenox was used for stroke prevention. However, when compared with clopidogrel in 20,332 patients with ischemic stroke,51 the rate of recurrent stroke with Aggrenox was similar to that with clopidogrel (9.0% and 8.8%, respectively) and the rate of major bleeding was higher with Aggrenox than with clopidogrel (4.1% and 3.6%, respectively). The results of this study dampened the enthusiasm for the use of Aggrenox. Because of its vasodilatory effects and the paucity of data supporting the use of dipyridamole in patients with symptomatic coronary artery disease, Aggrenox should not be used for stroke prevention in such patients.
Perioperative Management
Although dipyridamole has a rapid offset of action, the aspirin component of Aggrenox produces irreversible inhibition of COX-1. Consequently, as with aspirin, Aggrenox should be discontinued 7 to 10 days before surgery.
Glycoprotein IIb/IIIa Receptor Antagonists
As a class, parenteral GP-IIb/IIIa receptor antagonists have an established use in patients with acute coronary syndromes. The three agents in this class are abciximab, eptifibatide, and tirofiban.
Mechanism of Action
A member of the integrin family of adhesion receptors, GP-IIb/IIIa is found on the surface of platelets and megakaryocytes.52 With about 80,000 copies per platelet, GP-IIb/IIIa is the most abundant receptor. Consisting of a noncovalently linked heterodimer, GP-IIb/IIIa is inactive on resting platelets. When platelets are activated, inside-outside signal transduction pathways trigger a conformational activation of the receptor. Once activated, GP-IIb/IIIa binds adhesive molecules, such as fibrinogen and, under high shear conditions, vWF. Binding is mediated by the Arg-Gly-Asp (RGD) sequence found on the α chains of fibrinogen and on vWF, and by the Lys-Gly-Asp (KGD) sequence located within a unique dodecapeptide domain on the γ chains of fibrinogen. Once bound, fibrinogen and/or vWF bridge adjacent platelets together to induce platelet aggregation.52
Although abciximab, eptifibatide, and tirofiban all target the GP-IIb/IIIa receptor, they are structurally and pharmacologically distinct (Table 35-1). Abciximab is a Fab fragment of a humanized murine monoclonal antibody directed against the activated form of GP-IIb/IIIa.53 Abciximab binds to the activated receptor with high affinity and blocks the binding of adhesive molecules. In contrast to abciximab, eptifibatide and tirofiban are synthetic small molecules.53 Eptifibatide is a cyclic heptapeptide that binds GP-IIb/IIIa because it incorporates the KGD motif, whereas tirofiban is a nonpeptidic tyrosine derivative that acts as an RGD mimetic. Abciximab has a long half-life and can be detected on the surface of platelets for up to 2 weeks. Eptifibatide and tirofiban have shorter half-lives.53
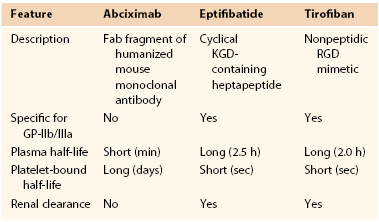
In addition to targeting the GP-IIb/IIIa receptor, abciximab also inhibits the closely related αvβ3 receptor, which binds vitronectin, and αMβ2, a leukocyte integrin. In contrast, eptifibatide and tirofiban are specific for GP-IIb/IIIa. Inhibition of αvβ3 and αMβ2 may endow abciximab with antiinflammatory and/or antiproliferative properties that extend beyond platelet inhibition.53
Dosing
All of the GP-IIb/IIIa antagonists are given as an intravenous bolus followed by an infusion. Because they are cleared by the kidneys, the doses of eptifibatide and tirofiban must be reduced in patients with renal insufficiency.53
Side Effects
Thrombocytopenia and bleeding are the most serious complications. Thrombocytopenia is immune-mediated and is caused by antibodies directed against neoantigens on GP-IIb/IIIa that are exposed upon antagonist binding.54 With abciximab, thrombocytopenia occurs in up to 5% of patients and is severe in about 1% of these individuals. Thrombocytopenia is less common with the other two agents, occurring in about 1% of patients.
Indications
Abciximab and eptifibatide are used in patients undergoing PCI, particularly those with acute MI.55 Tirofiban is used in high-risk patients with unstable angina. Eptifibatide also can be used for this indication.56
Perioperative Management
Abciximab binds GP-IIb/IIIa with higher affinity than eptifibatide or tirofiban, and it requires at least 12 hours after stopping the abciximab infusion for receptor occupancy to decline by 50%.57 This finding probably explains the higher transfusion requirement in patients taken to surgery less than 12 hours after abciximab discontinuation. Therefore abciximab should be stopped at least 12 hours before surgery. In contrast, because of their shorter half-lives, eptifibatide and tirofiban need only be stopped at least 2 hours preoperatively.58
New Antiplatelet Agents
Vorapaxar, an orally active inhibitor of the type 1 protease-activated receptor, the major thrombin receptor on platelets, has completed phase III clinical trials. When compared with placebo for secondary prevention in 26,449 patients with prior MI, ischemic stroke, or peripheral arterial disease, vorapaxar reduced the risk for the primary efficacy endpoint—the composite of cardiovascular death, MI, or stroke—by 13%, but doubled the risk for intracranial bleeding.59 In the prespecified subgroup of 17,779 patients with prior MI, however, vorapaxar reduced the primary efficacy endpoint by 20% (from 9.7% to 8.1%). Although the rate of intracranial hemorrhage in this group was higher with vorapaxar than with placebo (0.6% and 0.4%, respectively; P = 0.076) as was the rate of moderate or severe bleeding (3.4% and 2.1%, respectively; P < 00001), overall rates of bleeding were low. Based on these data, the drug is under consideration for regulatory approval in MI patients younger than the age of 75 years who weigh more than 60 kg and have no history of stroke or transient ischemic attack.
Anticoagulants
Anticoagulants are available in parenteral and oral forms. Currently available parenteral anticoagulants include heparin, low-molecular-weight heparin (LMWH), and fondaparinux, a synthetic pentasaccharide. Currently available oral anticoagulants include warfarin, dabigatran etexilate (an oral thrombin inhibitor), and rivaroxaban and apixaban, which are oral factor Xa inhibitors.
Parenteral Anticoagulants
Heparin
A sulfated polysaccharide, heparin is isolated from mammalian tissues rich in mast cells. Most commercial heparin is derived from porcine intestinal mucosa and is a polymer of alternating D-glucuronic acid and N-acetyl-D-glucosamine residues.60
Mechanism of Action.
Heparin acts as an anticoagulant by activating antithrombin (previously known as antithrombin III) and accelerating the rate at which antithrombin inhibits clotting enzymes, particularly thrombin and factor Xa.61 Antithrombin, the obligatory plasma cofactor for heparin,44,61 is a member of the serine protease inhibitor superfamily. Synthesized in the liver and circulating in plasma at a concentration of 2.6 ± 0.4 µM, antithrombin acts as a suicide substrate for its target enzymes.
To activate antithrombin, heparin binds to the serine protease inhibitor via a unique pentasaccharide sequence that is found on one third of the chains of commercial heparin (Fig. 35-4). The remainder of the heparin chains that lack this pentasaccharide sequence have little or no anticoagulant activity.62 Once bound to antithrombin, heparin induces a conformational change in the reactive center loop of antithrombin that renders it more readily accessible to its target proteases. This conformational change enhances the rate at which antithrombin inhibits factor Xa by at least two orders of magnitude, but has little effect on the rate of thrombin inhibition by antithrombin.63 To catalyze thrombin inhibition, heparin serves as a template that binds antithrombin and thrombin simultaneously.64 Formation of this ternary complex brings the enzyme in close apposition to the inhibitor, thereby promoting the formation of a stable covalent thrombin-antithrombin complex.64,65 Only pentasaccharide-containing heparin chains composed of at least 18 saccharide units (which corresponds to a molecular weight of 5400) are of sufficient length to bridge thrombin and antithrombin together.66 With a mean molecular weight of 15,000, and a range of 5000 to 30,000, almost all of the chains of unfractionated heparin are long enough to effect this bridging function.67 Consequently, by definition, heparin has equal capacity to promote the inhibition of thrombin and factor Xa by antithrombin and is assigned a 1 : 1 ratio of anti–factor Xa to anti–factor IIa (thrombin) activity.61
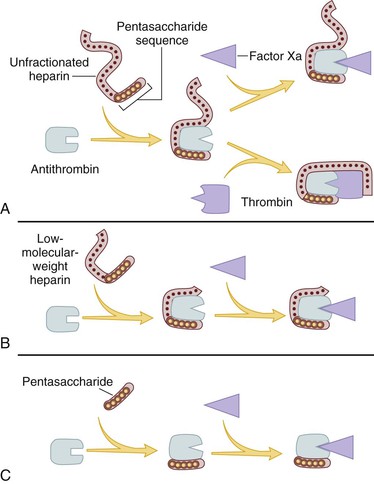
Figure 35-4 Mechanism of action of heparin, low-molecular-weight heparin (LMWH), and fondaparinux, a synthetic pentasaccharide. A, Heparin binds to antithrombin via its pentasaccharide sequence. This induces a conformational change in the reactive center loop of antithrombin that accelerates its interaction with factor Xa. To potentiate thrombin inhibition, heparin must simultaneously bind to antithrombin and thrombin. Only heparin chains composed of at least 18 saccharide units (corresponding to a molecular weight of 5400) are of sufficient length to perform this bridging function. With a mean molecular weight of 15,000, all of the heparin chains are long enough to do this. B, LMWH has greater capacity to potentiate factor Xa inhibition by antithrombin than thrombin because, with a mean molecular weight of 4500 to 5000, at least half of the LMWH chains are too short to bridge antithrombin to thrombin. C, Fondaparinux, a synthetic pentasaccharide, only accelerates factor Xa inhibition by antithrombin because the pentasaccharide is too short to bridge antithrombin to thrombin.
Heparin causes the release of tissue factor pathway inhibitor, a factor Xa–dependent inhibitor of tissue factor–bound factor VIIa,52 from the endothelium.67–69 Tissue factor pathway inhibitor may contribute to the antithrombotic activity of heparin. Longer heparin chains induce the release of more tissue factor pathway inhibitor than do shorter chains.67,68
Pharmacology of Heparin.
Heparin must be given parenterally. It is usually administered subcutaneously or by continuous intravenous infusion. When it is used for therapeutic purposes, the intravenous route is most often used. If heparin is given subcutaneously for treatment of thrombosis, the dose of heparin must be high enough to overcome the limited bioavailability associated with this method of delivery.61
In the circulation, heparin binds to the endothelium and to plasma proteins other than antithrombin. Heparin binding to endothelial cells explains its dose-dependent clearance. At low doses, the half-life of heparin is short because it rapidly binds to the endothelium.61 With higher doses of heparin, the half-life is longer because heparin is cleared more slowly once the endothelium is saturated.61 Clearance is mainly extrarenal; heparin binds to macrophages, which internalize and depolymerize the long heparin chains and secrete shorter chains back into the circulation. Because of its dose-dependent clearance mechanism, the plasma half-life of heparin ranges from 30 to 60 minutes with bolus intravenous doses of 25 and 100 units/kg, respectively.61
Once heparin enters the circulation, it binds to plasma proteins other than antithrombin, a phenomenon that reduces the anticoagulant activity of heparin.70 Some of the heparin-binding proteins found in plasma are acute-phase reactants whose levels are elevated in ill patients.70,71 Others, such as high-molecular-weight multimers of vWF, are released from activated platelets or endothelial cells.72,73 Activated platelets also release platelet factor 4 (PF4), a highly cationic protein that binds heparin with high affinity.74 The large amounts of PF4 found in the vicinity of platelet-rich arterial thrombi can neutralize the anticoagulant activity of heparin.75 This phenomenon may attenuate heparin’s capacity to suppress thrombus growth.
Because the levels of heparin-binding proteins in plasma vary from person to person, the anticoagulant response to fixed or weight-adjusted doses of heparin is unpredictable. Consequently, coagulation monitoring is essential to ensure that a therapeutic response is obtained. This is particularly important when heparin is administered for treatment of established thrombosis because a subtherapeutic anticoagulant response may render patients at risk for recurrent thrombosis,76,77 whereas excessive anticoagulation increases the risk for bleeding.78,79
The interaction of heparin with plasma proteins also contributes to the phenomenon of heparin rebound. Defined as the reappearance of anticoagulant activity after adequate neutralization of heparin with protamine, heparin rebound may contribute to excessive postoperative bleeding after cardiac surgery. This phenomenon likely reflects the slow release of protein-bound heparin after circulating heparin is neutralized by protamine and can be managed by administration of additional protamine as boluses or as a low-dose continuous infusion.80
Monitoring the Anticoagulant Effect of Heparin.
Heparin therapy can be monitored using the activated partial thromboplastin time (aPTT) or anti–factor Xa level. Although the aPTT is the test most often employed for this purpose, there are problems with this assay. aPTT reagents vary in their sensitivity to heparin and the type of coagulometer used for testing can influence the results.81 Consequently, laboratories must establish a therapeutic aPTT range with each reagent-coagulometer combination by measuring the aPTT and anti–factor Xa level in plasma samples collected from heparin-treated patients. For most of the aPTT reagents and coagulometers in current use, therapeutic heparin levels are achieved with a two- to threefold prolongation of the aPTT.82
Anti–factor Xa levels also can be used to monitor heparin therapy. With this test, therapeutic heparin levels range from 0.3 to 0.7 units/mL. Although this test is gaining in popularity, anti–factor Xa assays have yet to be standardized, and results can vary widely between laboratories.
Up to 25% of heparin-treated patients with venous thromboembolism require more than 35,000 units/day to achieve a therapeutic aPTT. These patients are considered heparin resistant.83 It is useful to measure anti–factor Xa levels in heparin-resistant patients because many will have a therapeutic anti–factor Xa level despite a subtherapeutic aPTT. This dissociation in test results occurs because elevated plasma levels of fibrinogen and factor VIII, both of which are acute-phase proteins, shorten the aPTT but have no effect on anti–factor Xa levels.84 Heparin therapy in patients who exhibit this phenomenon is best monitored using anti–factor Xa levels instead of the aPTT.83 Patients with congenital or acquired antithrombin deficiency and those with elevated levels of heparin-binding proteins also may need high doses of heparin to achieve a therapeutic aPTT or anti–factor Xa level. If there is good correlation between the aPTT and the anti–factor Xa levels, either test can be used to monitor heparin therapy.
Dosing.
For prophylaxis, heparin is usually given in fixed doses of 5000 units subcutaneously two or three times daily. With these low doses, coagulation monitoring is unnecessary.37 In contrast, monitoring is essential when the drug is given in therapeutic doses. Fixed-dose or weight-based heparin nomograms are used to standardize heparin dosing and to shorten the time required to achieve a therapeutic anticoagulant response. At least two heparin nomograms have been validated in patients with venous thromboembolism and reduce the time required to achieve a therapeutic aPTT.85,86 Weight-adjusted heparin nomograms also have been evaluated in patients with acute coronary syndromes. After an intravenous heparin bolus of 5000 units or 70 units/kg, a heparin infusion rate of 12 to 15 units/kg/hour usually is administered.86,87 In contrast, weight-adjusted heparin nomograms for patients with venous thromboembolism use an initial bolus of 5000 units or 80 units/kg followed by an infusion of 18 units/kg/hour.85 Thus patients with venous thromboembolism appear to require higher doses of heparin to achieve a therapeutic aPTT than do patients with acute coronary syndromes. This may reflect differences in the thrombus burden. Heparin binds to fibrin, and the fibrin content of extensive deep vein thrombi is greater than that of small coronary thrombi.
Limitations of Heparin.
Heparin has pharmacokinetic and biophysical limitations (Table 35-2). The pharmacokinetic limitations reflect heparin’s propensity to bind in a pentasaccharide-independent fashion to cells and plasma proteins. Heparin binding to endothelial cells explains its dose-dependent clearance, whereas binding to plasma proteins results in a variable anticoagulant response and can lead to heparin resistance.
Table 35-2
Pharmacokinetic and Biophysical Limitations of Heparin
| Limitations | Mechanism |
| Poor bioavailability at low doses | Binds to endothelial cells and macrophages |
| Dose-dependent clearance | Binds to endothelial cells and macrophages |
| Variable anticoagulant response | Binds to plasma proteins whose levels vary from patient to patient |
| Reduced activity in the vicinity of platelet-rich thrombi | Neutralized by platelet factor 4 released from activated platelets |
| Limited activity against factor Xa incorporated in the prothrombinase complex and thrombin bound to fibrin | Reduced capacity of heparin-antithrombin complex to inhibit factor Xa bound to activated platelets and thrombin bound to fibrin |
The biophysical limitations of heparin reflect the inability of the heparin-antithrombin complex to (a) inhibit factor Xa when it is incorporated into the prothrombinase complex,88,89 the complex that converts prothrombin to thrombin, and (b) to inhibit thrombin bound to fibrin.90,91 Consequently, factor Xa bound to activated platelets within platelet-rich thrombi has the potential to generate thrombin, even in the face of heparin.92 Once this thrombin binds to fibrin, it too is protected from inhibition by the heparin-antithrombin complex.93,94 Clot-associated thrombin can then trigger thrombus growth by locally activating platelets and amplifying its own generation through feedback activation of factors V, VIII, and XI.93–95 Further compounding the problem is the potential for heparin neutralization by the high concentrations of PF4 released from activated platelets within the platelet-rich thrombus.75
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


