Chapter 3 Anatomy, Physiology, and Pharmacology of the Vascular Wall
Normal Anatomy
The organization of the cellular elements and extracellular matrix components varies dramatically throughout the vasculature, accounting for its distinctive anatomic and physiologic features at various levels. Vessels larger than capillaries possess three distinct layers or tunics, called the intima, the media, and the adventitia. These layers are generally thicker and better defined in arteries than in veins. In arteries, the intima is composed of a sheet of endothelial cells lining the luminal surface and a subendothelial extracellular matrix. It is divided from the media by an internal elastic lamina. Rare inflammatory cells and smooth muscle cells may be found within the normal intima, although larger populations can be seen, generally as a reaction to injury or as a result of atherosclerotic disease. The media contains circular smooth muscle fibers embedded in a matrix of collagen, elastin, and proteoglycans and is divided from the adventitia by an external elastic lamina. Although the media is composed mostly of smooth muscle cells, there is increasing evidence that smooth muscle cell progenitors reside in niche populations within the media.1 Both the internal and external elastic laminae are visualized as bright white lines using B-mode ultrasonography, allowing a measurement of intima-media thickness, which can be used as a surrogate marker for atherosclerosis.2 The adventitia, which serves as the strength layer supporting endarterectomy, is composed primarily of loose connective tissue and fibroblasts. Inflammatory cells, nerve fibers, niche progenitor cells,1 and a nutrient microcirculation, known as the vasa vasorum,3 also reside within the adventitia.
Veins, as befitting their role as capacitance vessels under low-pressure conditions,4 are larger and thinner walled than arteries. The subendothelial layer of the intima is missing entirely, and an internal elastic lamina is apparent only in the larger veins. The medial layer contains few smooth muscle cells, collagen, and elastin. Thin bicuspid valves, consisting of two layers of endothelium sandwiched around a layer of connective tissue, are present in larger numbers in peripheral extremity veins, and rarely in central veins. The contractile state of both venules and veins is largely controlled by sympathetic adrenergic activity.
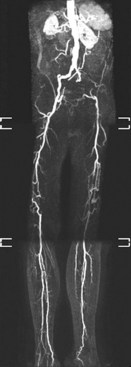
The arterial tree can be divided into three separate categories: large elastic arteries, medium muscular arteries, and small arteries. The aorta and its major branches are classified as large elastic arteries, the distributing arteries to major organs comprise the muscular arteries, and the arteries within organs compose the small arteries. From small arteries, blood flow travels through arterioles to capillary beds, postcapillary venules, and small veins and returns to the heart via larger veins. Collateral arteries are a special class of muscular arteries that traverse from one artery to another rather than feeding into arterioles. Normally there is little flow through collateral arteries and low shear stress. However, when the main conduit artery is obstructed, collateral artery flow and shear stress increase substantially as a compensatory mechanism, which after adaption eventually can restore up to one third of the normal conduit artery blood flow (Figure 3-1).
The aortic media is composed of well-defined lamellar units; each unit consists of a concentric plate of elastin and a circumferentially oriented layer of smooth muscle cells surrounded by a network of type III collagen fibrils embedded in a matrix of basal lamina. Finer elastin fibers compose a network between lamellae as do bundles of interstitial type I collagen.5 As the aorta traverses away from the heart, the percentage of collagen increases and that of elastin decreases, such that while the thoracic aorta and its major branches have more elastin than collagen, the abdominal aorta has more collagen than elastin. When thoracic aortic segments from multiple mammalian species across a wide range of body sizes were analyzed, the number of lamellar units was found to be proportional to the radius of the aorta, regardless of the wall thickness. The tangential tension on the artery wall can be roughly estimated with Laplace’s law (tension is proportional to the product of the radius and the pressure), which results in a remarkably constant average wall tangential tension per lamellar unit across different species.6 The upper two thirds of the thoracic aorta, which is thicker than 28 lamellar units, also contains a medial vasa vasorum.3 The dependence of the abdominal aortic wall on luminal nutrition may explain its increased propensity to aneurysm formation.
In contrast to elastic arteries where collagen and elastin comprise approximately 60% of the dry weight of the media, muscular arteries contain proportionally more smooth muscle cells and less collagen and elastin, allowing them to alter their diameter rapidly through vasodilation or vasoconstriction. In addition, their ratio of media to lumen is higher, contributing to their function as resistive arteries (Figure 3-2). Elastin is further lost as arteries become smaller, and the internal and external elastic lamellae become discontinuous and fragmented. The smallest arteries (arterioles) consist of only an endothelium, a layer of smooth muscle cells, and a filamentous collagenous adventitia. At the capillary level, only the endothelium remains, supported by an occasional contractile connective tissue cell known as a pericyte.7
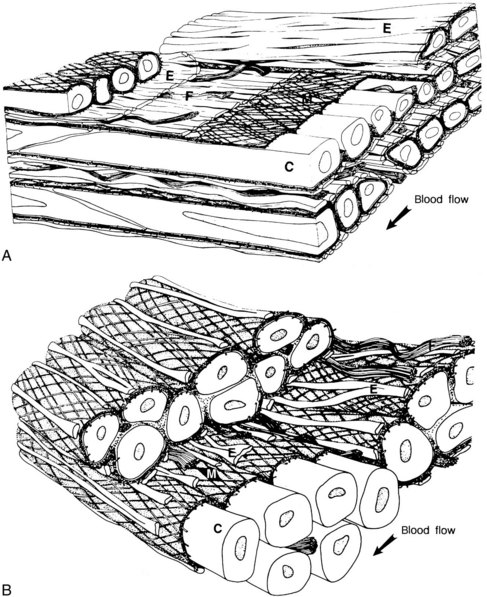
The differentiation of the three types of arteries has pathologic significance, as each class of vessel is subject to particular types of disease.8 Atherosclerosis affects the elastic and muscular arteries, whereas medial calcific sclerosis is confined to muscular arteries. Small arteries are subject to diffuse fibromuscular thickening and hyalinization.
Regulation of Luminal Area
Blood flow within the vasculature creates unique patterns of biomechanical forces on the vessel walls at different levels through the vascular tree. These biomechanical forces are pressure and shear stress. Pressure is created by the hydrostatic force created by cardiac contraction with the addition of the hydrostatic pressure created by gravity. It is a compressive force and also creates wall tension, as described by the law of Laplace. The greatest wall tension occurs in the large elastic vessels. Wall tension is distributed across all three layers of the vessel wall and determines wall thickness. Shear stress primarily affects the endothelium and is a result of drag caused by the tangential flow of viscous blood over the intimal surface. Endothelial cells align with the direction of this shear stress, which in laminar flow conditions is directly proportional to blood flow and fluid viscosity and inversely related to the cube of the radius.9 Shear stress is normally maintained in mammals at a constant between 10 and 20 dynes/cm2 at all levels of the arterial tree.10
Both developing and mature vessels respond to changes in hemodynamic forces by adjusting their diameters to maintain a constant level of shear stress. Acutely, this occurs by altering vasomotor tone.11 Vessels subjected to chronic changes in blood flow remodel by altering their structure in order to regain an appropriate level of shear stress and return vasomotor tone to normal.11 For example, during embryonic development, higher-volume flow leads to vessel enlargement, whereas lower-volume flow leads to vessel regression. Similarly, in adult vessels, an artery proximal to an arteriovenous fistula will enlarge and can eventually become aneurysmal.12 Conversely, an artery carrying less flow, either from proximal obstruction or a decrease in outflow (e.g., following amputation or paralysis) will adapt by decreasing its diameter.11,13,14
Diseased vessel segments also adapt to alterations in blood flow. A coronary artery with an enlarging atherosclerotic plaque is subject to increased blood flow velocity in the area of luminal stenosis. The coronary artery acutely responds by vasodilating and chronically undergoes a process known as outward remodeling to preserve luminal diameter (Glagov’s phenomenon). This adaptive process works to preserve a normal luminal diameter as long as the intimal lesion does not exceed 40% of the area within the internal elastic lamina, at which point pathologic narrowing begins.15 This process is dependent on an intact endothelium to translate the biomechanical information from shear stress to biochemical signals which regulate vessel diameter. Vessels denuded of endothelium in general do not respond to changes in flow.16
Pharmacologic agents regulating vasodilation and vasoconstriction affect vasomotor tone, and can be classified as endothelial and nonendothelial-dependent.17 Relaxation of the isolated rabbit aorta and other arteries induced by acetylcholine and other muscarinic receptor agonists was initially demonstrated to be dependent on the presence of endothelial cells by Furchgott and Zawadzki in 1980.18 In the absence of endothelial cells, acetylcholine causes contraction of the arterial wall instead of relaxation. In addition to acetylcholine, multiple other pharmacologic agents produce endothelial-dependent relaxation of vessels including arachidonic acid, adenosine triphosphate, adenosine diphosphate, bradykinin, histamine, norepinephrine, serotonin, thrombin, and vasopressin. The endothelial-dependence of these agents results from their ability to stimulate endothelial nitric oxide synthase (eNOS) to convert L-arginine to the soluble gas nitric oxide (NO; previously known as endothelial-derived relaxation factor and identified in 1987).19 NO stimulates guanylate cyclase in vascular smooth muscle cells leading to an increase in cyclic guanosine monophosphate and vasodilation. Nitric oxide is the most potent of the endothelial-derived relaxation factors; however, prostacyclin and endothelium-derived hyperpolarizing factors can also be demonstrated to be endothelial-derived vasodilators.20 In contrast, other pharmacologic agents such as adenosine, adenosine monophosphate, papaverine, isoproterenol, and nitrovasodilators (e.g., sodium nitroprusside) cause vasodilation even in the absence of endothelial cells.
In addition to releasing vasodilating factors, under different conditions the endothelium can also produce vasoconstricting factors in response to arachidonic acid, hypoxia, and in some isolated cerebral vessels by stretch.17,21 Arachidonic acid is metabolized by cyclooxygenase (COX) into endoperoxides, which are vasoconstrictors, and further metabolized by other enzymes to thromboxane, prostacyclin, and other prostaglandins, all of which can result in vasoconstriction. Reactive oxygen species, formed as by-products of COX generation of prostanoids, also stimulate vasoconstriction.21 Endothelin and angiotensin II are both peptide vasoconstrictors, which have been isolated from cultured endothelial cells. Increases in shear stress suppress endothelin gene expression and increase the production of eNOS by endothelial cells; endothelin promotes smooth muscle growth, whereas NO suppresses it.
It is likely that these endothelium-derived relaxing and constricting factors contribute to long-term vascular adaptation in response to changes in blood flow. Flow patterns also affect the expression of receptors involved in leukocyte recruitment, including intercellular adhesion molecule 1, vascular cell adhesion molecule 1, and monocyte chemoattractant protein-1.22 Missing or abnormal endothelium can contribute to certain pathologic conditions associated with acute and chronic vasospasm, such as atypical angina from coronary vasospasm and cerebrovasospasm after cerebral hemorrhage. In addition, endothelial dysfunction, as measured by abnormal flow-mediated vasodilation of the brachial or radial artery in response to reactive hyperemia, can be detected in the presence of most major cardiovascular risk factors, including hypertension, tobacco exposure (either active or passive), dyslipidemia, aging, diabetes mellitus, obesity, hyperhomocysteinemia, and chronic inflammation.20
Regulation of Medial and Intimal Thickening
As described previously, arterial wall thickness is initially determined by tangential tension. Wall thickening is a prominent feature of most pathologic processes. Hypertension causes arterial medial thickening in both humans and animals. Atherosclerosis, hypercholesterolemia, and reaction to injury such as endothelial denudation cause intimal thickening.23–26 Exactly how these responses are regulated is not clear, although it is certain that in each instance smooth muscle cells proliferate and drive accumulation of extracellular matrix.24,27 In addition, hypercholesterolemia promotes the accumulation of lipids and lipoproteins, followed by lipid-filled macrophages into the intimal lesion.24
Because smooth muscle accumulation is a central feature of most forms of vascular wall thickening, it is worth discussing the currently understood mechanisms of smooth muscle growth control.27,28 Although smooth muscle cell proliferation is an essential process during growth and development, these cells are predominantly quiescent in adult vessels. In the adult rat, smooth muscle cells turn over at a rate of 0.06% per day, which is barely detectable by available methods.29 Vascular smooth muscle cells can be stimulated by various pathologic conditions to undergo a phenotypic switch to a synthetic phenotype. In this state, they undergo high levels of proliferation, migration into the intimal layer, and generation of significant amounts of extracellular matrix components. A recent theory suggests that resident or perhaps bone marrow derived progenitor cells, rather than quiescent mature smooth muscle cells, are the primary cells contributing to vascular remodeling in response to arterial injury or disease.1,30 Regulation of the contractile and synthetic phenotypic states is incompletely understood, but appears to occur at the transcriptional level; this regulation clearly is critical to the problems of arterial wall remodeling in reaction to primary hypertension as well as local susceptibility to atherosclerotic change.
Because of the importance of vascular smooth muscle cells in pathologic processes, including atherosclerosis and restenosis in response to arterial and vein grafts as well as balloon angioplasty and stenting, many in vivo models of smooth muscle cell growth and proliferation have been developed. Perhaps increasingly relevant is a model using an angioplasty balloon catheter in the rat carotid artery, with or without stent implantation. In this model, significant intimal hyperplasia is only observed when the internal elastic lamina is ruptured.31,32 The best characterized model, however, is the balloon injury model, in which smooth muscle cell proliferation is stimulated by the passage of an inflated balloon catheter along an artery.33,34 The passage of the inflated balloon both stretches the arterial wall as well as denudes the endothelium. Immediately following balloon passage, platelets adhere to the denuded wall, spread, and degranulate, releasing numerous growth factors, chemotactic factors, and vasoactive substances.
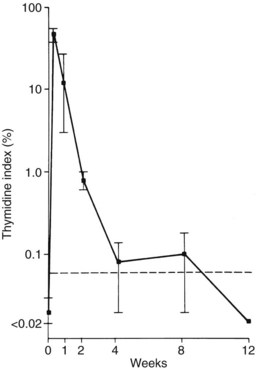
Endothelial denudation and platelet adherence are followed 1 to 2 days later by the stimulation of medial smooth muscle proliferation and migration across the internal elastic lamina to form a neointima.29 This response can be dramatic, as illustrated by the marked increase in thymidine incorporation index (a measure of DNA replication, and therefore proliferation) in the ballooned rat carotid artery (Figure 3-3). Smooth muscle cells commit to proliferation early after injury; this response can be blocked by giving heparin during the first few days after injury. Heparin blocks entry in the cell cycle, significantly reducing both the number of dividing cells and the eventual mass of neointima.35 Not all smooth muscle cells respond equally to mitogenic stimuli, probably because of the differing phenotypes of smooth muscle cells that can be derived from either the mesoderm or the ectoderm as well as the presence or absence of progenitor cells.30,36
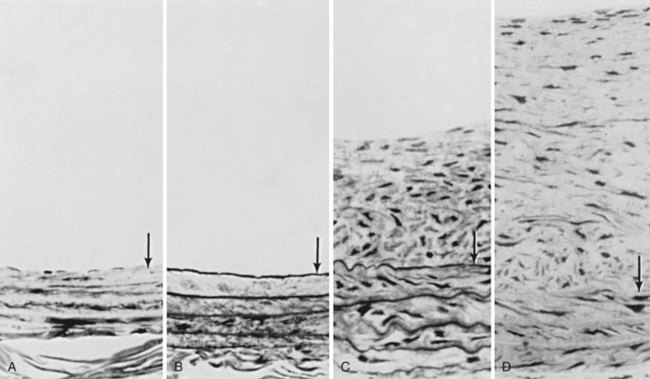
The initial proliferative response of smooth muscle cells occurs in the media of the injured artery and does not lead to an increase in wall thickness. Rather, the wall thickens only after the smooth muscle cells migrate into the intima and proliferate there. This process persists for approximately 2 weeks and spontaneously subsides regardless of whether endothelium reappears at the luminal surface. Intimal thickness is further increased by the accumulation of extracellular matrix synthesized by the smooth muscle cells (Figure 3-4).37
Several lines of evidence suggest that platelet degranulation stimulates smooth muscle cell proliferation and migration.25,38–42 Many growth factors, including platelet-derived growth factor (PDGF), transforming growth factor-β, and an epidermal growth factor–like protein, are found within platelet granules.38 Of these, PDGF appears to be the dominant growth factor affecting vascular smooth muscle cells after injury, as evidenced by work using anti-PDGF antibodies or infusion of PDGF after balloon injury.39,42 Injured arteries in thrombocytopenic animals show little intimal thickening.41 This effect occurs despite no measurable change in smooth muscle cell proliferation, suggesting that platelets are more important in stimulating migration rather than cell proliferation.40 However, it remains unknown where soluble growth factors and other platelet-derived granular proteins go after being released from the platelet. An attractive hypothesis is that they accumulate within the arterial wall and stimulate subsequent smooth muscle growth and migration.
Despite intensive study, the mechanisms that start or stop the intimal thickening process are still poorly understood. Several interesting and potentially important observations about the process have been made. First, the surface of the injured artery will only accumulate a single layer of platelets. Fibrin and microthrombi are seen at the luminal surface only when the artery that has already undergone intimal thickening is injured again or when small craters have been formed in the luminal surface in association with adherent macrophages in hypercholesterolemic animals.43 Therefore active fulminant thrombosis is not a usual reaction to injury; when it occurs, it must represent a major aberration of vessel function.
Second, in models in which reendothelialization occurs early or partial deendothelialization occurs without medial injury, intimal thickening does not develop although one or two rounds of medial smooth muscle cell proliferation may occur. This observation suggests that the endothelium normally suppresses smooth muscle proliferation and migration from the media into the intima. This suggestion is supported by the isolation of smooth muscle growth inhibitors from the vessel wall. In addition, the endothelium can synthesize a heparin-like molecule that inhibits in vitro smooth muscle cell growth; heparin suppresses both proliferation and migration of smooth muscle cells in vitro and in vivo.44 The endothelium also releases NO in a flow-dependent manner; NO is a growth inhibitor for smooth muscle cells.45,46 These findings suggest that an intact, functional endothelium actively maintains the medial smooth muscle cells and smooth muscle progenitor cells in a quiescent state instead of the quiescent state being attributable to the lack of growth factors. These findings also support the more general concept that the cells of the vascular wall communicate with each other and regulate each other’s function.
Third, the process of atherosclerosis appears to first require intimal thickening. The hypothesis that atherosclerosis results as a cellular response to endothelial injury was first proposed by Ross and Glomset in 1973.47 This theory has been modified and refined over the last 30 years, with a more recent recognition of the importance of the role of inflammation and inflammatory cells.48,49 In its current state, the theory proposes that injury leads to endothelial cell dysfunction, which changes endothelial permeability, adhesive characteristics, and responses to various growth factors. The changes in endothelial permeability allow inflammatory cells such as activated platelets, monocytes, and T lymphocytes to infiltrate into the arterial wall. The subsequent cell-cell interaction between endothelial cells, smooth muscle cells, and inflammatory cells create a fibroproliferative response, which eventually leads to the formation of atherosclerotic plaque.



