(1)
Division of Neonatal-Perinatal Medicine, Department of Pediatrics, Emory University, 2015 Uppergate Drive, Atlanta, GA 30322, USA
(2)
Children’s Healthcare of Atlanta Center for Developmental Lung Biology, Atlanta, GA, USA
(3)
Division of Neonatal-Perinatal Medicine, Department of Pediatrics, Emory University Emory + Children’s Healthcare of Atlanta Center for Developmental Lung Biology, Atlanta, GA, USA
Abstract
Individuals with alcohol use disorders (AUDs) have increased susceptibility to developing respiratory infections and the Acute Respiratory Distress Syndrome (ARDS). Oxidative stress is a primary contributor to the pathogenesis of these alcohol-related derangements. Sources of alcohol-induced lung oxidative stress include depletion of cytosolic and mitochondrial glutathione (GSH), increases in reactive oxygen species (ROS) and reactive nitrogen species (RNS), and enhanced expression and activity of NADPH oxidases (Nox). Alcohol-mediated oxidative stress in the lung leads to tissue injury and barrier dysfunction, phospholipid peroxidation and DNA oxidation, fibronectin production, apoptosis, and dysregulation of cellular zinc transport and immune function. Since these consequences directly relate to rising healthcare costs and associated hospitalizations of alcoholic patients, therapeutic interventions to attenuate alcohol-induced pulmonary oxidative stress are critical. Treatments recently under investigation in preclinical studies and, in some cases clinical studies, include drugs that activate peroxisome proliferator-activated receptor gamma (PPARγ), dietary zinc supplementation, and treatment with GSH precursors. These interventions are designed to attenuate alcohol-mediated increases in lung oxidative stress with the goal of restoring healthy lung function and thereby decreasing the risk of lung infections and injury in this vulnerable population.
Keywords
AlcoholLungOxidative stressRespiratory infectionsARDSAbbreviations
AUD
Alcohol use disorder
ARDS
Acute respiratory distress syndrome
GSH
Glutathione
ROS
Reactive oxygen species
RNS
Reactive nitrogen species
Nox
NADPH oxidases
PPARγ
Peroxisome proliferator-activated receptor gamma
•O2 −
Superoxide
H2O2
Hydrogen peroxide
•OH
Hydroxyl radical
•NO
Nitric oxide
ONOO-
Peroxynitrite
ELF
Epithelial lining fluid
GSSG
Glutathione disulfide
BAL
Bronchoalveolar lavage
TGFβ1
Transforming growth factor beta 1
SA
Small aggregate
LA
Large aggregate
MMP
Matrix metalloproteinase
TNFα
Tumor-necrosis factor alpha
GM-CSF
Granulocyte/macrophage colony-stimulating factor
ARE
Antioxidant response element
SAMe
S-Adenosyl-methionine
NAC
N-Acetylcysteine
GBS
Group B Streptococcus pneumoniae
Introduction
Individuals suffering from alcohol use disorders (AUDs) have a greater incidence of the Acute Respiratory Distress Syndrome (ARDS) compared to non-alcoholics [1]. During recent years, ample evidence has emerged describing the link between alcohol abuse and lung oxidative stress. Chronic alcohol consumption causes pulmonary oxidative stress through various mechanisms, including decreasing levels of the critical antioxidant glutathione (GSH), and renders the lung susceptible to injury [2]. Alcohol-induced oxidative stress affects whole lung tissue, multiple lung compartments such as epithelial lining fluid (ELF), and specific cell types such as epithelial cells, neutrophils, and alveolar macrophages. This chapter outlines recent investigations of the sources, consequences, and treatments of lung oxidative stress in the context of chronic alcohol use.
Sources of Alcohol-Induced Oxidative Stress
Chronic alcohol consumption increases lung oxidative stress. Alcohol-mediated pulmonary oxidative stress is generated from numerous sources, such as depletion of cytosolic and mitochondrial GSH, increases in reactive oxygen species (ROS) and reactive nitrogen species (RNS), and enhanced NADPH oxidases (Nox) expression and activity (Fig. 9.1). Several ROS produced in the lung include superoxide (•O2 −), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), and RNS that include nitric oxide (•NO), and peroxynitrite (ONOO−).
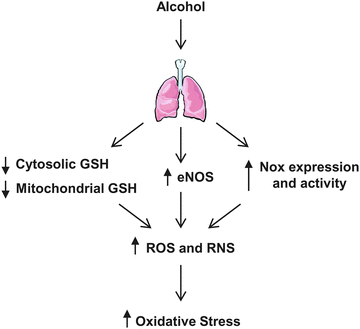
Fig. 9.1
Alcohol-induced sources of oxidative stress in the lung. Chronic alcohol use decreases cytosolic and mitochondrial GSH levels, and increases the expression and activity of endothelial nitric oxide synthase (eNOS) and NADPH oxidases (Nox) in the lung. These alcohol-induced alterations generate reactive oxygen species (ROS) and reactive nitrogen species (RNS)
In the lung, alcohol is metabolized by cytochrome p450 [3] into its major by-product acetaldehyde, which itself causes ROS production and lipid peroxidation [4]. When acetaldehyde is generated, antioxidants are utilized and depleted. GSH, the most abundant non-protein thiol in the body, is essential for cellular protection through detoxification of ROS. ARDS patients have decreased GSH levels in their ELF [5]. In fact, concentrations of oxidized GSH, known as glutathione disulfide (GSSG), were greater than the levels of GSH in the ELF of patients with ARDS [6]. Compared with non-alcoholic control subjects, ELF concentrations of GSH were significantly decreased in otherwise healthy chronic alcoholics (~580 μM in controls versus ~80 μM in alcoholics) [7]. Further, the percentage of GSSG was greater in chronic alcoholics (~10 % in alcoholics versus ~3 % in non-alcoholics) [7]. In a rat model of chronic alcohol consumption in which rats were fed normal chow but their drinking water contained alcohol (20 %) for >3 weeks, GSH levels were decreased in plasma, lung tissue, and lung lavage fluid, and GSSG levels were increased in lung lavage fluid [8]. Additionally, under normal conditions, GSH concentrations in the lung ELF are maintained at very high levels through active transport into this space by alveolar type II cells, with levels exceeding 400 μM, higher than the GSH levels in plasma or in other extracellular fluids [8]. Alcohol ingestion diminished GSH content in alveolar type II cells by 95 % [8] and contributed to alveolar epithelial barrier dysfunction [9]. Rats chronically fed alcohol additionally exhibited depleted GSH levels and increased ROS generation in the mitochondria of alveolar type II cells [2, 10, 11]. Dramatic decreases in GSH/GSSG ratios render the alveolar epithelium and the lung more susceptible to severe injury.
Critical sources of oxidative stress in the alcoholic lung are ROS and RNS. In rats fed liquid diets with or without alcohol (36 % of calories) for 6 weeks, •NO synthesis, metabolism, and release were determined in the lungs and in pulmonary microvascular endothelial cells. Compared to rats fed the control diet, alcohol-fed rats exhibited increases in lung H2O2 production, expression and activity of endothelial nitric oxide synthase (eNOS), and levels of protein nitration and oxidation [12]. Pulmonary microvascular endothelial cells from alcohol-fed rats had increased eNOS expression and activity [13] and •NO release [12]. In parallel, mice given alcohol (20 %) in their drinking water for 12 weeks also had increased expression of eNOS in their lungs [14]. Further, human umbilical vein endothelial cells exposed to 0.10 % alcohol in vitro for 3 days showed enhanced H2O2 production and eNOS expression [14]. These and other studies have identified that chronic alcohol consumption increases ROS and RNS production within the lung and thereby promotes oxidative and nitrosative stress.
NADPH oxidases (Noxes) are multicomponent, membrane-associated enzymes that utilize NADPH as an electron donor to catalyze the reduction of molecular oxygen to O2 •− and H2O2 [15]. Several Nox isoforms, specifically Nox1, Nox2, and Nox4, are expressed in the lung [16–18]. Studies in mice demonstrate that increased expression of either Nox1 or Nox2 is sufficient to up-regulate Nox4 expression [19]. In alcohol-exposed mouse embryos, Nox1, Nox2, and Nox4 constitute critical sources of ROS production [20]. Further, lungs from alcohol-fed mice showed increased expression and activity of Nox1 and Nox4 [14], and alveolar macrophages isolated from alcohol-fed mice demonstrated enhanced expression and activity of Nox1, Nox2, and Nox4, leading to increased ROS generation [19]. These studies show the critical role of Noxes in alcohol-induced reactive species production and collectively they implicate depletion of cytosolic and mitochondrial GSH, increases in reactive species, and enhanced NADPH oxidases (Nox) expression and activity, as key sources of pulmonary oxidative stress in the context of chronic alcohol use. The following section explores the pathological effects of chronic alcohol-mediated oxidative stress on the lung.
Consequences of Alcohol-Induced Oxidative Stress
Pulmonary oxidative stress induced by chronic alcohol consumption leads to a myriad of pathophysiological consequences. These consequences include lung injury and barrier dysfunction, phospholipid peroxidation and DNA oxidation, fibronectin production, apoptosis, and dysregulation of cellular zinc transport and immune function. As shown in Fig. 9.2, alcohol-induced oxidative stress leads to severe lung injury and immune dysfunction through increased protein concentrations in bronchoalveolar lavage (BAL) fluid, enhanced soluble e-selectin in plasma and ELF, decreased activity of surfactant phospholipids, increased DNA damage and protein oxidation, impaired tissue remodeling and repair, and immune cell dysregulation.
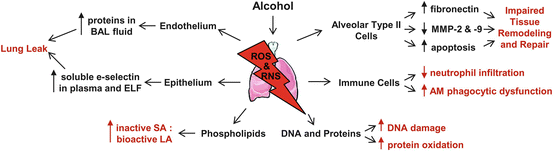
Fig. 9.2
Consequences of alcohol-induced oxidative stress in the lung. Chronic alcohol use primes the lung for severe injury and immune dysfunction. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) production induce pulmonary oxidative stress. Increased protein concentrations in bronchoalveolar lavage (BAL) fluid, enhanced soluble e-selectin in plasma and ELF, decreased active surfactant phospholipids, increased DNA damage and protein oxidation, impaired tissue remodeling and repair, and decreased neutrophil infiltration and alveolar macrophage (AM) function result from chronic alcohol-induced oxidative stress
Alcohol abuse itself does not cause acute lung injury; however, it renders the lung susceptible to dysfunction in response to inflammatory stimuli such as trauma, sepsis, or other clinical conditions that are recognized to cause ARDS [21]. Sepsis is commonly associated with the development of ARDS [22], and chronic alcohol abuse independently increases the incidence of ARDS in critically ill patients [23]. The incidence of ARDS in patients with a history of AUDs and sepsis combined is two- to four-times that of patients without a history of AUDs [21]. Additionally, patients with a history of AUDs and septic shock had more severe non-pulmonary organ dysfunction than non-alcoholics [24].
Alcohol abuse significantly increases the risk of sepsis-induced acute lung injury [25]. In experimental models, alcohol ingestion increases the expression of transforming growth factor beta 1 (TGFβ1) by activating the renin–angiotensin system that, through the actions of angiotensin II, induces oxidative stress and TGFβ1 expression that together contribute to alveolar epithelial barrier dysfunction, and these effects are exaggerated during endotoxemia [25]. The relatively permissive leak of proteins into the airspace of the alcoholic lung [14] not only contributes to the non-cardiogenic pulmonary edema that is the hallmark of ARDS, it may exacerbate the lung injury when these proteins become oxidized and interfere with normal alveolar functions.
Endothelial cell activation is also a critical step in the pathogenesis of ARDS. Soluble e-selectin is an endothelial cell-specific molecule that regulates leukocyte-endothelial cell adhesion and is important in endothelial cell and alveolar-capillary barrier function. Alcoholic patients with ARDS have increased concentrations of soluble e-selectin in both plasma and the ELF, which is consistent with endothelial cell and alveolar-capillary barrier dysfunction [26].
In addition, impaired lung surfactant production in alcoholics [8, 9, 27] is proposed to contribute to their increased susceptibility to ARDS. In the ELF of ARDS patients, there are increased ratios of inactive small aggregate (SA) surfactant phospholipids to bioactive large aggregate (LA) surfactant phospholipids. In an experimental model, chronic alcohol ingestion in rats prior to inducing acute sepsis via cecal ligation and perforation increased lung lavage protein levels, aggravated hypoxemia, and attenuated the pool of functional LA surfactant phospholipids [22].
Chronic oxidative stress induced by alcohol exposure also has detrimental effects on lung DNA and proteins including fibronectin, a matrix glycoprotein implicated in lung injury and repair. In the ELF and alveolar macrophages isolated from the lungs of patients with a history of AUDs, alcohol abuse increased the expression of fibronectin. Alcohol also attenuated the expression of matrix metalloproteinase (MMP)-2 and -9 [29], which are implicated in tissue remodeling. Further, alveolar type II cells isolated from ethanol-fed rats showed increased oxidative stress and fibronectin expression, leading to subsequent lung tissue remodeling and stimulation of pro-inflammatory mediators that prime the alcoholic lung to injury [30]. Alcohol-induced oxidative stress also mediates dysfunction in immune cells, such as neutrophils, monocytes, and alveolar macrophages. In rats administered alcohol (4 g/kg body weight as a 50 % solution) by gastric gavage, there was increased DNA damage and protein oxidation in the lung tissue [28].
Since alveolar type II cell viability is critical for epithelial repair, the effect of chronic alcohol ingestion on the sensitivity of alveolar type II cells to inflammatory mediators that are up-regulated during sepsis is very important. Alveolar epithelial type II cells isolated from alcohol-fed rats have decreased mitochondrial GSH levels along with increased susceptibility to apoptosis and necrosis [11]. Further, chronic alcohol exposure exacerbates the oxidative stress caused by either tumor-necrosis factor alpha (TNFα) or H2O2 treatment of rat alveolar type II cells alone via mitochondrial GSH depletion and apoptosis through caspase-3 activation [10, 11]. Overall, there is compelling evidence that chronic alcohol ingestion sensitizes alveolar type II cells to inflammatory mediator-induced apoptosis and necrosis, thereby impairing the ability of these cells to promote repair following damage to the alveolar epithelium.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


