Acute Heart Failure
Srinivas Iyengar
Garrie J. Haas
James B. Young
Overview
Since the early 1980s, hospital discharges for heart failure (HF) have increased more than 150% and now represent the most common reason for hospitalization in the elderly (1). The incidence of HF approaches 10 per 1,000 population after the age of 65 years, and it was estimated that in 2005 alone, the direct and indirect costs of managing this disease process in the United States approached 28 billion dollars. This considerable economic burden is fueled by the astonishingly high early hospital readmission rate for patients with HF. It is estimated that nearly 50% of patients initially admitted with decompensated HF are readmitted within 60 to 90 days after hospital discharge (2). A seeming paradox is that most patients with HF do not die in a hospital. Although in-hospital mortality for acute HF averages 5% to 8%, the 1-year mortality following an acute HF exacerbation requiring hospitalization may be as high as 60%. Given these staggering statistics, it is essential for the clinician to recognize this syndrome early and to institute appropriate therapy in a timely fashion. Clinicians should focus on both the rapid reversal of HF symptoms and the appropriate transition to accepted long-term outpatient HF therapies.
Acutely decompensated HF (ADHF) is a complex syndrome that is most often a direct result of myocardial injury caused by underlying ischemia or infarction. Although coronary artery disease (CAD) is the most common cause of acute HF in Western societies, a broad spectrum of other potential causes exists, including such diverse conditions as acute valvular abnormalities, pericardial disease, and acute myocarditis (Table 86.1). Hypertension and HF resulting from diastolic dysfunction may precipitate as many as 50% of HF-related hospitalizations in the elderly, particularly in women, a situation not previously generally recognized (3). The management of acute HF requires rapid identification of the underlying cause, with specific treatment directed at reversing the associated abnormal pathophysiologic state. The support of patients while the diagnostic evaluation is pursued can range from urgent therapy with intravenous vasodilators and diuretics to more aggressive pharmacologic support with varying combinations of inotropic, vasodilator, and vasopressor agents. In complicated or confusing situations, hemodynamic monitoring is essential. When a reparable lesion has been identified or when cardiac transplantation is considered an option, mechanical circulatory support may be required until definitive treatment can be instituted. Because of the complexity and diverse nature of the acute HF syndrome, treatment must be individualized. Finally, unfortunately, it must be pointed out that for many different reasons, few short-term or long-term large-scale, properly powered morbidity and mortality trials have been completed in patients with acute HF or ADHF. Perhaps this explains why consensus cannot be often reached regarding the best management strategies with few definitive guidelines published.
Patients presenting with acute HF may be classified into one of three fundamental categories, based in part, on differences in pathophysiology and the chronicity of their cardiac disease (2). These categories of acute HF are new-onset HF, decompensation of previously stable chronic HF (ADHF), and advanced or “end-stage” HF that has responded poorly to standard medical therapy with a downward spiral of symptoms and physical findings.
The syndrome of new-onset HF, particularly when it is acute in presentation, represents a true medical emergency that warrants an expedient diagnostic and therapeutic approach. If appropriate treatment is not instituted within a reasonable period, irreversible cardiac decompensation may ensue, leading to a progressive syndrome of shock, multiorgan failure, and death (Fig. 86.1). New-onset acute HF is generally recognized when symptoms develop within hours to days in patients without a history of prior cardiac decompensation (4). A history of previous treatment for heart disease may be elicited because many patients will have experienced symptoms (i.e., angina or exertional dyspnea) related to myocardial ischemia. In fact, atherosclerotic heart disease and the complications of myocardial ischemia and infarction are the most common causes of acute HF in Western societies, though other conditions can precipitate this condition. Many of the conditions resulting in new-onset HF may be definitively treated, thus resulting in a favorable impact on the patient’s short- and long-term course. Often, a rapid decision regarding the appropriateness of surgical intervention is necessary if the patient is to realize a favorable outcome. For example, the 1-year mortality rate for patients presenting with acute pulmonary edema, a common
clinical manifestation of new-onset HF, may exceed 50% when an underlying lesion or condition is not defined and treated (5). In certain life-threatening presentations, such as acute ischemic myocardial injury or complications thereof (i.e., acute valvular regurgitation, ventricular septal defect, cardiac tamponade), immediate evaluation and directed therapy are indicated for stabilization and treatment of the acute HF syndrome. In these emergency situations, definitive therapy should not be delayed by prolonged efforts to achieve a more stable clinical situation. Without correction of the underlying lesions, such efforts are often unsuccessful and commonly result in further clinical deterioration with the development of irreversible myocardial dysfunction. When the disorder precipitating new-onset HF does not appear to be one requiring urgent therapy, definitive diagnostic studies and consideration of specific forms of therapy can be delayed until clinical and hemodynamic stabilization has been achieved.
clinical manifestation of new-onset HF, may exceed 50% when an underlying lesion or condition is not defined and treated (5). In certain life-threatening presentations, such as acute ischemic myocardial injury or complications thereof (i.e., acute valvular regurgitation, ventricular septal defect, cardiac tamponade), immediate evaluation and directed therapy are indicated for stabilization and treatment of the acute HF syndrome. In these emergency situations, definitive therapy should not be delayed by prolonged efforts to achieve a more stable clinical situation. Without correction of the underlying lesions, such efforts are often unsuccessful and commonly result in further clinical deterioration with the development of irreversible myocardial dysfunction. When the disorder precipitating new-onset HF does not appear to be one requiring urgent therapy, definitive diagnostic studies and consideration of specific forms of therapy can be delayed until clinical and hemodynamic stabilization has been achieved.
TABLE 86.1 Major Causes of Acute Heart Failure | |
|---|---|
|
Patients with advanced or end-stage HF comprise only about 2% of all HF hospital admissions (6). These patients typically present with symptoms of congestion, poor peripheral perfusion, and multiorgan dysfunction, and they are often cachectic and functionally extremely limited. They have usually failed over a long period to respond to standard medical, surgical, and device therapies, and their prognosis without extreme measures is exceedingly poor. These are the patients who are classified in American College of Cardiology/American Heart Association (ACC/AHA) stage D category (Fig. 86.2; see also Chapter 87), and they may be considered and evaluated for advanced surgical therapies including cardiac transplantation and so-called destination therapy with mechanical assist devices (the destination is the patient’s home). If these options are not tenable, then palliative therapy and hospice referral should be considered.
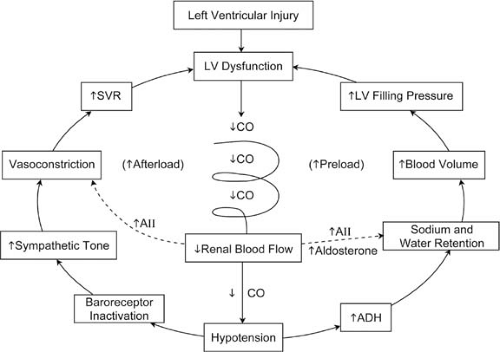 FIGURE 86.1. Depicted are the factors in cardiogenic shock perpetuating continued left ventricular (LV) dysfunction and progressive deterioration in cardiac output (CO). As CO falls, renal blood flow is reduced, which thus activates the renin-angiotensin-aldosterone system. Angiotensin II (AII) directly results in vasoconstriction and indirectly increases blood volume by enhancing aldosterone release. Hypotension resulting from a further fall in CO activates the central nervous system release of antidiuretic hormone (ADH). Inactivation of baroreceptors enhances sympathetic tone and vasoconstriction. As CO, renal blood flow, and hypotension remain reduced, preload and afterload continue to increase, and this leads to further LV dysfunction. ↑, increased; ↓, decreased; SVR, systemic vascular resistance. |
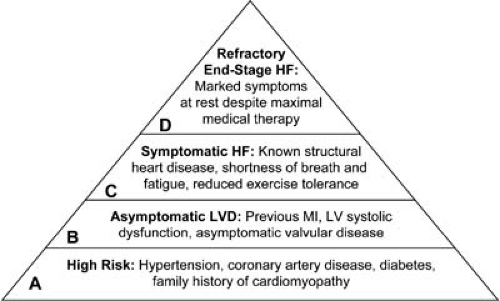 FIGURE 86.2. Disease progression of heart failure (HF): American College of Cardiology/American Heart Association HF stages. LV, left ventricular; LVD, left ventricular dysfunction; MI, myocardial infarction. (Adapted from Hunt, SA, Baker, DW, Chin, MH, et al. 2001 ACC/AHA guidelines for the evaluation and management of chronic heart failure in the adult: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines Committee to Revise the 1995 Guidelines for the Evaluation and Management of Heart Failure: developed in collaboration with the International Society for Heart and Lung Transplantation, endorsed by the Heart Failure Society of America. Circulation 2001;104:2996–3007 .) |
A prime focus of this chapter is the most common reason for HF hospitalization, acute decompensation in the patient
with previously stable HF (ADHF). Approximately 75% of HF admissions fall into this category, and unlike the conditions discussed earlier, medical management is usually the preferred therapeutic approach. Distinct clinical findings differentiating those with ADHF from new-onset and stable chronic HF are presented in Table 86.2. These patients comprise ACC/AHA stage C HF and often have a history of one or more prior hospitalizations for ADHF. These patients are usually congested with normal peripheral perfusion at presentation, and they generally respond favorably to the rapid institution of vasoactive therapy of one sort or another.
with previously stable HF (ADHF). Approximately 75% of HF admissions fall into this category, and unlike the conditions discussed earlier, medical management is usually the preferred therapeutic approach. Distinct clinical findings differentiating those with ADHF from new-onset and stable chronic HF are presented in Table 86.2. These patients comprise ACC/AHA stage C HF and often have a history of one or more prior hospitalizations for ADHF. These patients are usually congested with normal peripheral perfusion at presentation, and they generally respond favorably to the rapid institution of vasoactive therapy of one sort or another.
TABLE 86.2 Comparison of Acute and Chronic Heart Failure | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Historical Perspective
Significant advances in the pharmacologic support of ADHF have been made over the past 4 decades. In the 1960s, only digitalis and diuretics were available for HF management, and furosemide, approved for use in 1967, was the mainstay of drug therapy for ADHF. The intravenous inotropic, vasodilator, and vasopressor therapies introduced in the 1970s proved to be effective in the stabilization of many patients presenting with ADHF. Newer forms of these medications that are currently available and/or being studied are a testament to the role these drugs have assumed in correcting the underlying hemodynamic derangements seen in ADHF. Important progress has been made toward the development of novel, more directed therapies with less toxic side effects. At the same time, much has been learned regarding important limitations of other vasoactive therapies, particularly those that stimulate the myocardium and increase inotropy. No longer is the so-called dobutamine “holiday” advocated as a “tune-up” for mildly decompensated patients, given the substantive toxicities of inotropic support. As with the pharmacologic approach for treating chronic HF, the treatment of ADHF has also recently been influenced by our newer understanding of the HF neurohormonal pathophysiologic paradigm. Unfortunately, to date there have been few randomized, controlled clinical trials in ADHF management, and thus specific evidence-based guidelines are not generally available. Only the recently released European Society of Cardiology HF Guidelines attempt to address this patient population, and these recommendations are hampered by absence of reliable clinical trial data and consensus of clinicians (especially those with ADHF treatment expertise in North America). Thus, the management of ADHF is less standardized and highly variable. Recently, the development of large acute HF registries such as the Acute Decompensated Heart Failure National Registry (ADHERE Registry) (7) has provided important observational data regarding HF demographics and therapy in more than 100,000 patients. These data promise to advance our understanding of the ADHF syndrome significantly. Certainly, ongoing clinical trials in ADHF will have a major impact in the future as standardized and evidence-based methods of managing these patients are developed.
Pathophysiology
Acute decompensation in a patient with previously documented chronic HF represents a different pathophysiologic state than that of the acute HF syndrome in patients without prior left ventricular dysfunction or those with chronic HF. Patients with ADHF have a marked reduction in left ventricular systolic or diastolic function at baseline caused by a previous substantial injury to the myocardium. In approximately 50% of cases, the underlying disease is ischemic in origin. In the remainder, various causes may be identified, such as valvular disease or long-standing hypertension. Most cases of nonischemic chronic HF are idiopathic (8). In most instances, systolic and diastolic abnormalities are present. The typical patient with chronic HF is likely receiving some combination of oral therapy (angiotensin-converting enzyme [ACE] inhibitors or angiotensin-receptor blockers [ARBs], β-blockers, aldosterone antagonists, vasodilators, diuretics, digoxin) in an attempt to maintain stability and adequate functional capacity. Despite this treatment, however, these patients generally have mild to moderate symptoms of HF on a long-term basis and may exhibit some degree of volume overload even in the compensated state. Left ventricular wall stress, because of left ventricular dilatation, is usually elevated, and compensatory neurohormonal
mechanisms are activated (see Chapter 85). When acute decompensation occurs, these baseline abnormalities become further deranged. This is associated with a further deterioration in hemodynamic status that manifests primarily as an increase in left ventricular filling pressure. Further neurohormonal perturbation ensues, including activation of the renin-angiotensin-aldosterone axis, sympathetic nervous system, arginine vasopressin, and endothelin, leading to systemic and pulmonary vasoconstriction with sodium and water retention. Although initially compensatory, this process becomes maladaptive over time and causes disease progression. In a minority of patients with ADHF, cardiac output is reduced on presentation and is seen clinically as multiorgan hypoperfusion. Renal function must be monitored closely because the cardiorenal syndrome, reflecting poor renal perfusion with rising serum creatinine, is increasingly recognized as a predictor of poor outcome during ADHF hospitalization (9,10). The worsening of congestion and increasing left ventricular filling pressures in the patient with ADHF may lead to a progression of subendocardial ischemia, atrioventricular valvular regurgitation, alterations in extracellular matrix deposition and repair, and continued detrimental left ventricular remodeling (11). This process leads to progressive myocardial dysfunction, thereby increasing the risk of recurrent ADHF (12). In addition, the arrhythmia threshold is lowered as a result of this process, and the risk of sudden cardiac death is thus increased. Given these potential adverse effects of congestion, it is not surprising that persistent elevation of left ventricular filling pressures in patients with ADHF is an important independent predictor of survival, and the reduction of these pressures remains a major goal of pharmacologic therapy (13) (Fig. 86.3).
mechanisms are activated (see Chapter 85). When acute decompensation occurs, these baseline abnormalities become further deranged. This is associated with a further deterioration in hemodynamic status that manifests primarily as an increase in left ventricular filling pressure. Further neurohormonal perturbation ensues, including activation of the renin-angiotensin-aldosterone axis, sympathetic nervous system, arginine vasopressin, and endothelin, leading to systemic and pulmonary vasoconstriction with sodium and water retention. Although initially compensatory, this process becomes maladaptive over time and causes disease progression. In a minority of patients with ADHF, cardiac output is reduced on presentation and is seen clinically as multiorgan hypoperfusion. Renal function must be monitored closely because the cardiorenal syndrome, reflecting poor renal perfusion with rising serum creatinine, is increasingly recognized as a predictor of poor outcome during ADHF hospitalization (9,10). The worsening of congestion and increasing left ventricular filling pressures in the patient with ADHF may lead to a progression of subendocardial ischemia, atrioventricular valvular regurgitation, alterations in extracellular matrix deposition and repair, and continued detrimental left ventricular remodeling (11). This process leads to progressive myocardial dysfunction, thereby increasing the risk of recurrent ADHF (12). In addition, the arrhythmia threshold is lowered as a result of this process, and the risk of sudden cardiac death is thus increased. Given these potential adverse effects of congestion, it is not surprising that persistent elevation of left ventricular filling pressures in patients with ADHF is an important independent predictor of survival, and the reduction of these pressures remains a major goal of pharmacologic therapy (13) (Fig. 86.3).
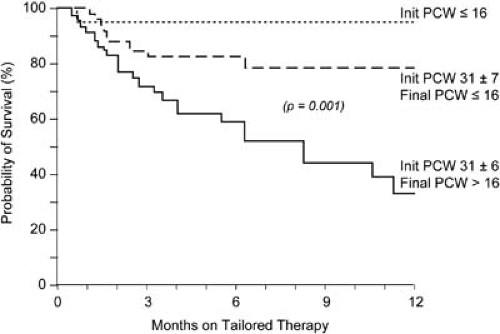 FIGURE 86.3. Relationship between pulmonary capillary wedge pressure (PCWP) and survival in 152 patients referred for heart transplantation with ejection fractions lower than 20% and receiving optimal medical therapy. Init, initial. (Adapted from Stevenson LW, Tillisch JH, Hamilton M, et al. Importance of hemodynamic response to therapy in predicting survival with ejection fraction <20% secondary to ischemic or non-ischemic dilated cardiomyopathy. Am J Cardiol 1990;6:1348–1354 .) |
Presentation and Evaluation
When patients first present to the emergency department or outpatient office with signs and symptoms suggesting ADHF, a methodical and rapid assessment should ensue. Patients with ADHF usually present with congestive symptoms and preserved cardiac output. They may describe worsening exertional dyspnea or dyspnea at rest, orthopnea, paroxysmal nocturnal dyspnea, and fatigue. According to the ADHERE registry, the most common presenting symptom is dyspnea, which occurs in 89% of patients, whereas fatigue (an indication of low cardiac output) is much less common, occurring in only 32% (Table 86.3). Because symptoms of dyspnea and edema have a broad differential diagnosis, particularly in the elderly and in those patients with multiple comorbid conditions, additional testing may be necessary before a firm diagnosis can be made. The history should explore possible precipitating factors for ADHF. These factors are presented in Table 86.4 and include such diverse causes as dietary and medical noncompliance, arrhythmias, and drugs such as nonsteroidal antiinflammatory agents. New or recurrent ischemia or infarction is a serious and potentially life-threatening cause of decompensation and should be considered in all patients even if they were previously determined to have “normal” coronary arteries.
TABLE 86.3 Clinical Presentation of Patients Hospitalized with Heart Failure | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
In conjunction with the history, the patient should have a targeted physical examination with emphasis on the presence of jugular venous distention, abnormal heart and lung sounds, displaced apical impulse, and evidence of fluid retention. Evidence of low cardiac output (tachycardia, hypotension, cool extremities, mental status changes) should also be thoroughly evaluated. In most patients with ADHF, findings of the physical examination often reflect volume overload with peripheral edema and jugular venous distention. Pulmonary rales may be present but are certainly not as impressive as in the patient with acute myocardial injury and pulmonary edema. In fact, pulmonary findings may be relatively unremarkable in patients with ADHF even when cardiac filling pressure is markedly elevated. In the ADHERE registry, only 68% of patients admitted
with ADHF had pulmonary rales (Table 86.3). A gallop rhythm and murmur of mitral regurgitation are typically audible, and the cardiac impulse is enlarged, sustained, and laterally displaced (14). Certain findings on physical examination are prognostically important. Jugular venous distention and an S3 gallop have been reported to be independent predictors of hospitalization and all-cause mortality (15). A broad range of variability exists concerning physical findings, which often depend on the underlying lesion and duration of cardiac dysfunction.
with ADHF had pulmonary rales (Table 86.3). A gallop rhythm and murmur of mitral regurgitation are typically audible, and the cardiac impulse is enlarged, sustained, and laterally displaced (14). Certain findings on physical examination are prognostically important. Jugular venous distention and an S3 gallop have been reported to be independent predictors of hospitalization and all-cause mortality (15). A broad range of variability exists concerning physical findings, which often depend on the underlying lesion and duration of cardiac dysfunction.
TABLE 86.4 Common Causes of Acute Decompensated Heart Failure in Patients with Chronic Heart Failure | |
|---|---|
|
A laboratory evaluation including complete blood count, serum electrolytes, glucose, blood urea nitrogen, creatinine, and liver function tests should all be part of the initial diagnostic protocol for a patient presenting with ADHF. Thyroid function tests and cardiac biomarkers can be performed if one suspects an underlying endocrine or ischemic condition that precipitated the event. Anemia and poorly controlled diabetes may also cause or complicate ADHF. As discussed previously, the finding of renal impairment has important implications regarding risk stratification and prognosis. The idea of risk stratification for patients with ADHF has recently come to light from an additional analysis of the ADHERE database (16). Utilizing a classification and regression tree (CART) analysis, this inquiry found that patients admitted to the hospital for ADHF could be stratified into low-, intermediate-, and high-risk groups for in-hospital mortality by means of routine blood tests and vital signs. The single best predictor for mortality was a high blood urea nitrogen level (>43 mg/dL), followed by a low systolic blood pressure on admission (<115 mm HG), and then by an elevated serum creatinine level (>2.75 mg/dL). These results are informative because they provide clinical parameters in which risk assessment can be made on an immediate basis by the clinician.
Types A, B, and C natriuretic peptides (NPs) appear to be beneficial counterregulatory hormones produced by the heart (types A and B) or the vascular endothelium (type C) in response to certain pathophysiologic events. Indeed, these hormones may play a significant role in attenuating potentially detrimental left ventricular hypertrophy, and they protect the heart during ischemic events (17,18,19). BNP is an endogenously secreted hormone constitutively released by the heart under conditions of increased pressure or volume overload (20). BNP has been found to have vasodilatory and natriuretic properties, as well as potentially mediating the effects of the renin-angiotensin-aldosterone pathway (21). Although several blood assays for natriuretic peptides have been developed, arguably the most experience exists with determination of BNP levels. Measurement of plasma BNP is recommended when the diagnosis based on history and physical examination remains uncertain. The introduction of a rapid, point-of-care assay method of measuring BNP levels has facilitated the use of this test in the emergency department and outpatient setting. The Breathing Not Properly (BNP) study was a prospective study of 1,586 patients who came to the emergency department with complaints of acute dyspnea (22) and who had a BNP level drawn on presentation. It was reported that BNP levels greater than 100 pg/mL were diagnostic of HF with a sensitivity of 90%, a specificity of 76%, and a predictive accuracy of 83%. The BNP test is best utilized when integrated into the entire clinical evaluation. Relying on a single BNP measurement to diagnose ADHF, in the absence of a thoughtful and complete clinical evaluation, should be discouraged. BNP levels can vary with age (increased) and sex (higher in women) and can be elevated in renal insufficiency, which is most likely the result of decreased clearance. Morbid obesity can also affect interpretation of the BNP level, resulting in a falsely low level. Because of the high negative predictive value of this test, we have found the BNP test to be most helpful when the results are low (<100 pg/mL) in a dyspneic patient in whom the cause of the problem is uncertain. In this setting, ADHF as a cause of dyspnea can be excluded with a high degree of certainty. Figure 86.4 incorporates the BNP test into a diagnostic algorithm for the patient presenting with dyspnea.
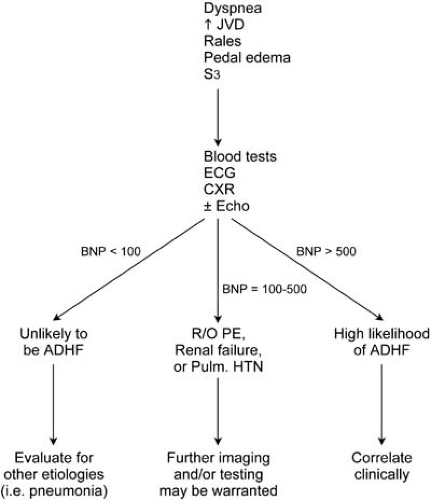 FIGURE 86.4. Ladder diagram for clinical stratification according to brain natriuretic peptide (BNP) levels. Emphasis should be placed on the history and physical examination, along with correlation with the appropriate diagnostic testing. ADHF, acutely decompensated heart failure; CXR, chest radiograph; ECG, electrocardiogram; Echo, echocardiogram; HTN, hypertension; JVD, jugular venous distention; PE, pulmonary embolism; R/O, rule out. |
A chest radiograph (CXR) provides a rapid method of visualization of the heart as well the lung spaces. The presence of cardiomegaly and/or infiltrative processes in the lung fields can help better direct the clinician toward a pulmonary versus cardiac disorder. In the ADHERE database, 75% of patients admitted with ADHF had radiographic evidence of pulmonary congestion, whereas 25% had no such finding, again reinforcing the poor predictive value of a single test such as a CXR for diagnosing ADHF.
An electrocardiogram should be done to evaluate for any underlying ischemic event. Rhythm disorders such as atrial fibrillation and complete heart block can result in the signs and symptoms of ADHF. Identification of pacemaker dysfunction may also be important, particularly in the current era of cardiac resynchronization and biventricular pacing therapy for HF.
Echocardiography is an excellent tool for evaluating certain parameters including wall motion abnormalities, valvular function, and the presence of myocardial infiltrative processes. Ejection fraction and left ventricular dimensions determined from an echocardiogram have prognostic implications. All patients who have a new diagnosis of HF should have an echocardiogram as part of their initial workup. If a patient with a history of chronic HF presents with an episode of acute decompensation, we believe that another echocardiogram should be performed at the clinician’s discretion.
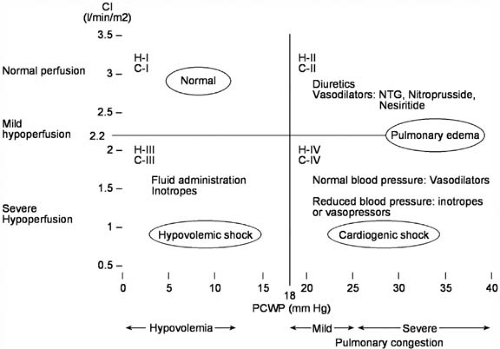 FIGURE 86.5. Four-square diagram of hemodynamic assessment of patients with heart failure (Forrester classification). H-I to H-IV refers to hemodynamic severity, with cardiac index (CI) and pulmonary capillary wedge pressure (PCWP) shown on the vertical and horizontal axes, respectively. C-I to C-IV refers to clinical severity. NTG, nitroglycerin. (Adapted from Forrester JS, Diamond GA, Swan HJ. Correlative classification of clinical and hemodynamic function after acute myocardial infarction. Am J Cardiol 1977;39:137–145 .) |
Classification of Hemodynamic Profile
Following the initial clinical evaluation of the patient with ADHF, a decision must be made regarding the therapeutic approach. Those patients with evidence of myocardial ischemia, significant volume overload, severe symptoms of dyspnea, significant renal insufficiency, hypotension, or other major comorbid conditions complicating their HF syndrome should be hospitalized for aggressive vasoactive and diuretic therapy. In some cases when symptoms and volume excess are mild, the patient may be treated in the emergency department or observation unit with close follow-up in the outpatient setting. The use of a “four-square” model (Fig. 86.5) can help to identify specific hemodynamic profiles in the patient with ADHF quickly and can aid the clinician in instituting therapy expeditiously. This diagram, initially proposed by Forrester et al. in 1977 for the clinical assessment of patients after myocardial infarction, was modified in 1999 by Lynne Warner Stevenson, now of the Brigham and Women’s Hospital and Harvard University in Boston, to characterize patients admitted with ADHF better and to facilitate communication regarding prognosis and therapies for ADHF (23) (Fig. 86.6). Most patients admitted with ADHF are in profile B (“wet and warm”), whereas a much smaller number is in profile C (“wet and cold”). Hemodynamic profile L (“cold and dry”) is rare and typically results from overdiuresis of a profile C patient. The prognosis is better for a profile B patient compared with profile C; the prognosis for profile L patients is unknown, but it is probably better than for profile C. Patients admitted with dyspnea who are subsequently found to have normal filling pressures (not congested) and a normal cardiac output are in profile A (“warm and dry”), which is the hemodynamic goal for all patients. In profile A patients who are hospitalized for dyspnea or edema, symptoms may be caused by conditions other than chronic HF, such as chronic pulmonary disease, arrhythmia, or transient ischemia associated with valvular dysfunction (24). The ability to determine the appropriate hemodynamic profile for any patient with ADHF is based on an accurate initial evaluation, as previously outlined.
Pharmacotherapeutics
Goal of Acute Pharmacologic Therapy
The objectives of the pharmacologic management of ADHF are to relieve disabling symptoms and discomfort rapidly, to reverse hemodynamic derangement, to preserve myocardial blood flow and energetics, to prevent end-organ dysfunction, and to stabilize the patient while further definitive diagnostic evaluation and therapy are pursued (Table 86.5). Of course, keeping the patient alive during the hospital admission is paramount, but, as noted previously, most patients succumb to HF in a nonacute hospital setting. Acute intravenous therapy is directed toward improving overall ventricular performance by favorably affecting the major determinants of ventricular function, including ventricular preload, afterload, and myocardial
contractility. As discussed previously, the ability to reduce pulmonary congestion and left ventricular filling pressure in the acute setting has important prognostic implications and is a major goal of ADHF therapy. Dyspnea in this setting, after all, is clearly related to elevated left ventricular end-diastolic pressure and pulmonary hypertension. The medications commonly used to accomplish this goal are generally administered intravenously, exhibit pharmacokinetic properties that allow for rapid titration to hemodynamic effect, and have a relatively short plasma half-life, so any untoward effect can be quickly terminated (Table 86.6). The medications commonly used to initially treat the ADHF patient are discussed in detail.
contractility. As discussed previously, the ability to reduce pulmonary congestion and left ventricular filling pressure in the acute setting has important prognostic implications and is a major goal of ADHF therapy. Dyspnea in this setting, after all, is clearly related to elevated left ventricular end-diastolic pressure and pulmonary hypertension. The medications commonly used to accomplish this goal are generally administered intravenously, exhibit pharmacokinetic properties that allow for rapid titration to hemodynamic effect, and have a relatively short plasma half-life, so any untoward effect can be quickly terminated (Table 86.6). The medications commonly used to initially treat the ADHF patient are discussed in detail.
 FIGURE 86.6. Modified four-square diagram of hemodynamic assessment of patients with heart failure (HF). This diagram emphasizes the bedside presentation of patients with acutely decompensated HF. ACE, angiotensin-converting enzyme; JVD, jugular venous distention; PND, paroxysmal nocturnal dyspnea. (Adapted from Stevenson LW. Tailored therapy to hemodynamic goals for advanced heart failure. Eur J Heart Fail 1999;1:251–257 .) |
TABLE 86.5 Goals for Patients Hospitalized with Heart Failure | |
|---|---|
|
Acute Vasodilator Therapy
Nitroglycerin
Nitroglycerin is often utilized in the early management of ADHF for patients fitting the warm and wet hemodynamic profile (profile B). The major hemodynamic effect of nitrates is to evoke a reduction in ventricular filling pressure and volume by increasing venous capacitance through venodilation (25,26,27,28,29,30,31,32,33,34,35). This shifts central blood volume to the venous capacitance vessels, which decreases pulmonary congestion and improves the symptoms of breathlessness associated with ADHF and elevated ventricular filling pressures. This vasodilating action is mediated through relaxation of vascular smooth muscle resulting primarily from a nitrate-induced production of S-nitrosothiols, which cause an increase in intracellular cyclic guanosine monophosphate (cGMP) (36,37). Nitrates may also elicit vasodilation by other mechanisms, including stimulation of the endothelial production and release of prostacyclin or prostaglandin E (38,39,40). Although these agents are predominantly considered for venodilation, higher doses of nitrates produce arterial dilatation and therefore reduce systemic vascular resistance (SVR) and ventricular impedance, particularly in the setting of systemic vasoconstriction (41).
Nitrates are generally effective in improving dyspnea in patients with ADHF, and therapy can be instituted rapidly when it is evident that pulmonary congestion is present and requires preload reduction. Intravenous nitroglycerin is the preparation most commonly used. However, sublingual tablets and a lingual-buccal spray may occasionally be used until an intravenous preparation is available to be infused (42,43). Intravenous nitroglycerin (typically starting at a dose of 0.2 μg/kg per minute) may be rapidly titrated upward in increments of 0.1 to 0.2 μg/kg per minute to improve symptoms and to reduce pulmonary and systemic venous pressures and resistance, ventricular filling pressures, and, to a variable degree, SVR. The ability of nitrates to augment stroke volume and cardiac output by decreasing left ventricular afterload is often dose dependent and usually requires an initial intravenous dose of at least 0.4 μg/kg per minute (27). Interestingly, the only major randomized controlled trial to date assessing the efficacy of nitroglycerin for use in ADHF was the Vasodilation in the Management of Acute Congestive Heart Failure (VMAC) trial, which showed that nitroglycerin was less effective than nesiritide and not different than placebo in improving a number of clinical and hemodynamic parameters (44) (see the later discussion of nesiritide).
One must also consider the propensity for the development of pharmacodynamic tolerance during continuous intravenous infusions, which necessitates an intermittent increase in dose to maintain the desired hemodynamic effect (45,46). In the VMAC trial, those patients who underwent hemodynamic monitoring with a pulmonary artery catheter received higher doses of nitroglycerin than patients in whom hemodynamics were estimated clinically. In those monitored with a pulmonary artery catheter, there was a progressive escalation of nitroglycerin dose to achieve favorable hemodynamics over time. Another problem with infusion of nitroglycerin intravenously is that polyvinyl chloride tubing and solution bags adsorb the drug. These observations suggest that the development of nitrate tolerance and the need to escalate dosing are critical issues, and these problems may go unrecognized in the clinical setting when invasive monitoring is not utilized (44).
TABLE 86.6 Intravenous Agents for Acute Decompensated Heart Failure | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Nitroprusside
Sodium nitroprusside is a powerful venous and arterial vasodilator with potent afterload-reducing properties (47). Nitroprusside relaxes arterial and venous smooth muscle via the production of nitric oxide and nitrosothiols, leading to an increase in cGMP and smooth muscle relaxation (29). As with nitroglycerin, nitroprusside causes preload reduction by diminishing heightened venous tone and increasing venous capacitance with a concomitant shift in central blood volume to the periphery (30). This causes a reduction in right ventricular pressure and volume. More relevant to nitroprusside is its rapid and powerful effect on afterload. This agent reduces the major components of aortic impedance (mean and hydraulic vascular load), and the results are improved and often dramatic increases in forward stroke volume and cardiac output with reductions in left ventricular filling pressure, volume, and valvular regurgitation (25,27,30,48,49,50,51). Fortunately, this decrease in afterload occurs without major changes in aortic pressure (49). This characteristic of nitroprusside enhances its efficacy in patients fitting the wet and cold profile (profile C) when the reduction in stroke volume is secondary to a high SVR.
In most patients with ADHF, judicious titration of nitroprusside can result in decreased aortic impedance, increased cardiac output, and reduced ventricular filling pressures without the undesirable effects of a decrease in systemic blood pressure or rise in heart rate. Unlike nitroglycerin, nitroprusside may cause “coronary steal” whereby arteriolar dilatation in nonischemic zones diverts coronary flow away from areas of ischemia (52,53). The frequency with which this occurs in HF is not well documented.
Nitroprusside is administered intravenously with an infusion pump and is usually initiated at a dose of 0.10 to 0.20 μg/kg per minute. Its effects are evident within 60 to 90 seconds after initiation of the infusion, and should an adverse effect such as symptomatic hypotension occur, the vasodilating properties usually abate within 20 to 30 minutes after discontinuation (25). In ADHF, insertion of an arterial catheter for continuous systemic blood pressure recording and monitoring and for frequent blood gas determinations is often recommended. One should recognize, however, that during nitroprusside infusion, the pressure measured in a peripheral artery (i.e., radial artery) may not reflect a reduction in central aortic pressure because of nitroprusside-induced changes in the amplitude and timing of reflected waves within the central aorta (54). One must remain cognizant of this when the clinical findings are consistent with systemic hypoperfusion despite a seemingly acceptable peripheral arterial pressure.
Nitroprusside can be rapidly titrated to achieve the desired clinical and hemodynamic end points, including a reduction in pulmonary capillary wedge pressure (PCWP) to 18 to 20 mm Hg, a decrease in SVR to 1,000 to 1,200 dynes/cm5 per second, a reduction in valvular regurgitation, and an improvement in stroke volume, cardiac output, and systemic perfusion, while avoiding significant hypotension and tachycardia (Fig. 86.7). In ADHF, particularly when myocardial ischemia is present,
attention to Starling mechanisms with respect to preload and augmentation of stroke volume remains important. Although nitroprusside is titrated to achieve hemodynamic goals, rarely are doses greater than 4 to 5μg/kg per minute required to maintain adequate vasodilation in the acute HF setting, and doses this high for prolonged periods (>72 hours) should generally be avoided because of the risk of thiocyanate and cyanide toxicity (55,56).
attention to Starling mechanisms with respect to preload and augmentation of stroke volume remains important. Although nitroprusside is titrated to achieve hemodynamic goals, rarely are doses greater than 4 to 5μg/kg per minute required to maintain adequate vasodilation in the acute HF setting, and doses this high for prolonged periods (>72 hours) should generally be avoided because of the risk of thiocyanate and cyanide toxicity (55,56).
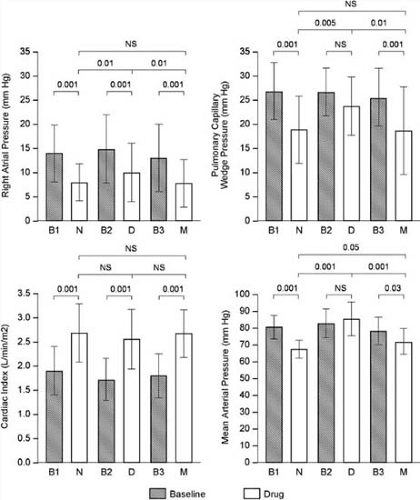 FIGURE 86.7. Effects of sodium nitroprusside (N), dobutamine hydrochloride (D), and milrinone lactate (M) on right atrial pressure, pulmonary capillary wedge pressure, cardiac index, and mean arterial pressure in patients with severe heart failure (B1, B2, and B3 represent baseline measurements). At dosages evoking a similar increase in cardiac index, all agents significantly reduced right atrial pressure, with nitroprusside and milrinone also producing a significant reduction in mean arterial pressure compared with baseline. The effect of milrinone and nitroprusside on pulmonary capillary wedge pressure was greater than that observed with dobutamine. NS, not significant. (Adapted from Monrad ES, Baim DS, Smith HS, et al. Milrinone, dobutamine, and nitroprusside: comparative effects on hemodynamics and myocardial energetics in patients with severe congestive heart failure. Circulation 1986;73[suppl III]:III168–III174. ) |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


