Abnormalities of the Aortic Root
Matina Prapa
John Pepper
Michael A. Gatzoulis
Introduction
Progressive aortic root dilation has been reported in a number of congenital heart defects, both repaired and unrepaired (1). Following extensive research in this field over the past few decades, the thoracic aortic wall is no longer considered as a simple tube but rather a highly sophisticated structure responsive to local hemodynamic changes (2). Inherited connective tissue disorders, such as Marfan disease and bicuspid aortic valve (BAV), have served as aortopathy models, both in terms of pathogenesis and medical management. However, similar aortic wall abnormalities have been reported in a wider range of disparate congenital heart defects, including coarctation of the aorta, tetralogy of Fallot (TOF), and complete transposition of the great arteries (TGA) (Table 34.1). Aortic dilation is usually silent but may produce symptoms from ensuing progressive aortic regurgitation or acute aortic dissection. Thus, early identification of aortopathy is important for appropriate follow-up and timely intervention. As life expectancy of patients with congenital heart disease (CHD) increases, several cardiovascular risk factors superimpose on inherent wall abnormalities, and may increase the incidence of aortic complications. The following sections will review the most prevalent CHD lesions associated with aortic root abnormalities and the underlying pathology, diagnostic evaluation, and treatment options.
Marfan Syndrome
Marfan syndrome (MFS) is a heritable connective tissue disorder resulting from mutations in the fibrillin-1 (FBN1) gene located on chromosome 15 (15q21.1). The estimated prevalence of MFS is 1 in 5,000 to 10,000 live births, affecting both sexes equally (13). Up to 95% of individuals fulfilling the clinical criteria for MFS have an FBN1 mutation (14). Approximately a quarter of affected individuals have de novo mutations, with the absence of family history of the disease. MFS has an autosomal dominant pattern of inheritance with a high penetrance, affecting almost all allele carriers, but marked phenotypic variability. The syndrome has several manifestations from the skin and the skeletal, cardiovascular, and ocular systems and is considered a prototype model for aortic pathology; practically all affected patients present with aortic dilation or dissection during their lifetime (15). Underlying histologic abnormalities of the aneurysmal aortic wall have been described applying the term “cystic medial necrosis,” characterized by elastic fiber fragmentation, noninflammatory loss of vascular smooth muscle cells (VSMCs), and accumulation of basophilic ground substance within cell-depleted areas in the aortic media (13). However, as described later, these changes are not unique to MFS but can be found in other causes of nonsyndromic thoracic aortic aneurysms (TAAs) (16).
TABLE 34.1 Commonest Congenital Heart Disease Lesions Associated with Aortic Root Abnormalities | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Despite the identified causative role of FBN1 mutations, the exact sequence of molecular changes leading to MFS continues to be under investigation. FBN1 is a main structural component of the extracellular microfibrillar network that connects VSMCs to the surrounding elastic fibers, facilitating tissue elasticity (17). At present, more than 1,000 mutations have been described in the FBN1 gene without a clear genotype–phenotype association (13). Mutations in exons 24–32 may be indicators of earlier onset of morbidity in MFS although the latter were not associated with a higher risk of aortic dissection in patients with aortic dilation (18). Due to the established weakness of connective tissue, early theories on the pathogenesis of MFS focused on the structural properties of FBN1. A dominant negative effect was initially suggested, where the mutant allele gives rise to abnormal FBN1 that in turn, disrupts the assembly of normal fibrillin polymers (19,20). However, a different study in transgenic mice has exhibited that the mutated protein participates in productive microfibrillar formation (18). Moreover, the addition of a normal allele in the same animal model rescued the aortic phenotype, highlighting the contribution of haploinsufficiency to the disease (21). This raises the possibility that the normal aortic wall lamellar structure can be restored.
More recently, the importance of transforming growth factor beta (TGFβ) in the pathogenesis of MFS has emerged. The TGFβ family of cytokines plays a central role in vascular remodeling and aneurysm formation (2). Mouse models of the disease have shed light on the regulatory functions of FBN1 which controls the bioavailability of TGFβ by binding a latent form of the cytokine. Excessive TGFβ signaling has been shown to play a causal role in aortic root enlargement, mitral valve abnormalities, and emphysema associated with MFS (22,23,24). More importantly, the administration of TGFβ-neutralizing antibody has lead to reversal of the above manifestations (13). The general mechanistic hypothesis for the above findings is that FBN1 deficiency makes sequestered TGFβ more accessible to activation, although mutated forms of the protein can also stimulate TGFβ release (25). The significance of the TGFβ pathway was confirmed with the discovery of a second locus for MFS, termed as MFS type II (MFS2), associated with mutations in the TGFβ type II receptor (TGFBR2) (26). Overall, it is hypothesized that FBN1 deficiency results in altered homeostasis of the extracellular matrix and subsequent increase of TGFβ activity. The exact sequence of succeeding mechanisms leading to aortic wall degeneration remains elusive. However, it has been established that increased elastolysis occurs, with activation of matrix metalloproteinases (MMP-2 and MMP-9), apoptosis of VSMC, and abnormal cell migration (27).
Diagnosis
The diagnosis of MFS is largely based on clinical grounds. Due to the wide range of clinical manifestations, a multidisciplinary approach is required. A definite diagnosis can be made applying the Ghent criteria, which are based on demonstration of symptoms from different organ systems and family history of MFS (13). However, Ghent nosology cannot exclude MFS in children, due to the variability in onset and severity of symptoms in this age group (28). Consequently, long-term follow-up is required in younger patients before a diagnosis can be established. Recently, an international expert board has published a revised Ghent nosology, in which aortic root aneurysm and ectopia lentis are the cardinal features of MFS with a new scoring system for other systemic features of the disease (Table 34.2) (29). Genetic screening can
be adjunctive to clinical evaluation, in cases with suspected MFS or where other family members carry an identified causal mutation. However, FBN1 mutations are not specific to MFS and can be found in a wide range of phenotypes including mitral valve, aorta, skeleton, and skin (MASS) syndrome, familial mitral valve prolapse, and familial ectopia lentis (26). Mutations in TGFBR2 can be found in up to 21% of MFS patients who are FBN1 negative (30,31). Conversely, the above type of the disorder, termed as MFS type 2, has a phenotype overlapping with that of Loeys–Dietz syndrome (15). The presence of ectopia lentis, which is absent in the latter, can be useful in the differential diagnosis between the two syndromes (32).
be adjunctive to clinical evaluation, in cases with suspected MFS or where other family members carry an identified causal mutation. However, FBN1 mutations are not specific to MFS and can be found in a wide range of phenotypes including mitral valve, aorta, skeleton, and skin (MASS) syndrome, familial mitral valve prolapse, and familial ectopia lentis (26). Mutations in TGFBR2 can be found in up to 21% of MFS patients who are FBN1 negative (30,31). Conversely, the above type of the disorder, termed as MFS type 2, has a phenotype overlapping with that of Loeys–Dietz syndrome (15). The presence of ectopia lentis, which is absent in the latter, can be useful in the differential diagnosis between the two syndromes (32).
TABLE 34.2 Revised Ghent Nosology for the Diagnosis of Marfan Syndrome | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
The most characteristic and troublesome features of MFS involve the cardiovascular system and include TAA formation, aortic regurgitation owing to an enlarged aortic root, and mitral valve prolapse (15). Aortic enlargement is typically located at the level of sinuses of Valsalva and may eventually extend to the sinotubular junction and proximal ascending aorta (Fig. 34.1B) (33). Approximately 35% of MFS patients develop aortic root dilation by the age of 5 years and 68% by the age of 19 years (3). Dilation is progressive although its rate is unpredictable and can vary even in the same individual (34). The incidence of serious cardiovascular complications, including aortic dissection or rupture, has been estimated to be around 4.3% in childhood with a rise to approximately 20% in adolescence (4). Aortic regurgitation may develop in 15% to 44% of MFS patients during childhood and adolescence, and has been strongly associated with acute cardiovascular events (35). Additional predictors of aortic complications include younger age at presentation and a family history of severe aortic disease (4). Patients diagnosed with MFS during childhood have significantly fewer adverse cardiac events compared to those diagnosed during adulthood, highlighting the importance of early identification of the disease (36).
Following diagnosis of MFS, an echocardiogram is recommended at 6 months to assess the progression of aortic disease. If measurements remain stable, echocardiographic follow-up can be performed on an annual basis (15). However, those guidelines refer to adult patients and it has been suggested that younger patients should be monitored every 6 months, particularly during accelerated growth phases (37). Children who do not meet the full diagnostic criteria of the disease should be screened at least every 5 years, until they reach adulthood (37). Baseline echocardiographic imaging requires diameter measurement at the level of the sinuses of Valsalva, which is the typical affected aortic segment in MFS. Of note, aortic root diameters should be indexed to age and body surface area (38). It has been suggested that an adjusted nomogram with higher upper limits should be used for the diagnosis of aortic dilation in children with MFS, as they tend to be taller individuals compared to their peers (39). In the instance of poor echocardiographic windows, magnetic resonance imaging (MRI) or computed
tomography (CT) angiograms should be performed (Fig. 34.2A). Importantly, both of the above modalities measure external diameter, which is 0.2 to 0.4 cm higher compared to the internal diameter measured by echocardiography (15). Precise measurements are difficult due to movement of the ascending aorta in three planes and the difficulty of obtaining a true cross-sectional diameter of the aorta. Therefore, recommendations in asymptomatic patients based on diameter values in guidelines should be made with caution.
tomography (CT) angiograms should be performed (Fig. 34.2A). Importantly, both of the above modalities measure external diameter, which is 0.2 to 0.4 cm higher compared to the internal diameter measured by echocardiography (15). Precise measurements are difficult due to movement of the ascending aorta in three planes and the difficulty of obtaining a true cross-sectional diameter of the aorta. Therefore, recommendations in asymptomatic patients based on diameter values in guidelines should be made with caution.
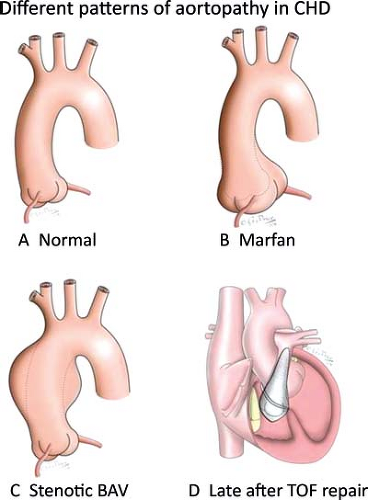 Figure 34.1 Different patterns of aortopathy seen in congenital heart disease (CHD). A: Anterior view of a normal aortic root, ascending aorta, and aortic arch. B: Typical location of aortic dilation at the level of sinuses of Valsalva in patients with Marfan syndrome. C: Asymmetric pattern of ascending aortic dilation encountered in stenotic bicuspid aortic valve (BAV) disease. D: Uniform dilation of aortic root and ascending aorta following repair of tetralogy of Fallot (TOF). |
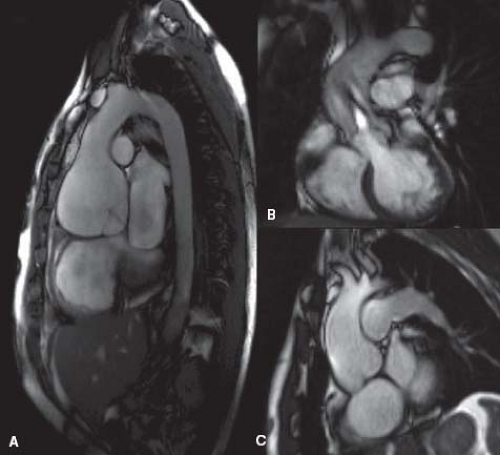 Figure 34.2 Magnetic resonance images of different types of aortic dilation. A: Still frame from steady state free precession image showing the “pear-shaped” aortic dilation seen in Marfan syndrome. A jet of mild aortic regurgitation is seen. B: Jet lesion directed to the anterior-lateral portion of the ascending aorta in a stenotic bicuspid aortic valve with asymmetrical dilation of the vessel wall at the same level. C: Dilatation of the aortic root extending to the sinotubular junction and proximal ascending aorta in a patient with repaired tetralogy of Fallot. |
Routine CT or MRI imaging of the entire thoracic aorta is recommended in patients who present with descending aortic dilation, type B aortic dissection, or following ascending aortic aneurysm repair (40). The vast majority of MFS patients present with dilation of the ascending aorta or type A dissection (15). However, in a smaller subset of patients, the disease initially involves the distal thoracic aorta with reported poor outcomes in cases of acute type B aortic dissection (41). Moreover, up to 52.6% of MFS patients who underwent initial repair of the ascending aorta had subsequent vascular procedures involving portions of their descending thoracic aortas (42), prompting comprehensive imaging follow-up. Increased regional wall stiffness assessed by MRI may be an additional marker for extensive screening, as it has been shown to be a predictor of progressive descending aortic dilation in MFS (43).
Therapy
Both medical and surgical treatments have substantially increased life expectancy in MFS, extending it up to 70 years compared to 40 years in the 1970s (42). Conservative treatment consists of use of β-blockers and exercise limitation, with an aim to reduce arterial stress in MFS. β-blockade may be initiated at the time of diagnosis or in the presence of significant aortic dilation (40). Dosage must be closely monitored during accelerated growth phases and adjusted in case of adverse effects, such as asthma aggravation. If not well tolerated, medical treatment can be substituted with a calcium channel blocker. A protective effect of both agents has been reported on aortic growth rate in children and adolescents with MFS (44).
The therapeutic benefit of β-blockers may be secondary to negative chronotropy and inotropy resulting in decreased arterial stress (40). Another antihypertensive drug, the angiotensin II type 1 receptor (AT1) blocker, losartan has been shown to improve the aortic phenotype in FBN1-deficient mice (24). Following establishment of aortic root aneurysm at 7 weeks of age, 6-month treatment with losartan prevented elastic fragmentation and reduced TGFβ signaling in the aortic media, compared to propranolol, which beneficial effect was restricted to reduction of aortic root growth rate (24). In a pilot study including 18 children with MFS, the use of angiotensin II–receptor blockers (ARBs) significantly slowed the rate of progressive aortic root dilation (45). Subsequently, the beneficial additive effect of losartan to β-blockade in retardation of aortic dilation has been exhibited in a small pilot study in 28 MFS children and young adults as well as the much larger COMPARE trial including 233 MFS adults (46,47). However, in a much anticipated trial comparing losartan with atenolol in 608 MFS patients from 21 clinical centers, there was no significant difference in the rate of aortic root dilation between the two groups over a period of 3 years (48). This lack of superiority may lead to rejection of losartan as an alternative first-line treatment option, although some critical points to be taken into consideration when interpreting the above results include the lack of a placebo arm and the advanced aortic disease of study participants compared to the early developmental stages of the equivalent mouse models benefiting from losartan (24,48).
Despite long-term treatment, major cardiovascular complications may still develop (44). Therefore, pharmacologic therapy can be used to delay surgical treatment in affected children but the need for close follow-up is fundamental. Avoidance of maximal and isometric exertion is also recommended, although exercise restrictions may not always be applicable in younger children. An American
Heart Association consensus document published in 2004 offers recommendations for physical activity in young patients with genetic cardiovascular diseases, including MFS (49).
Heart Association consensus document published in 2004 offers recommendations for physical activity in young patients with genetic cardiovascular diseases, including MFS (49).
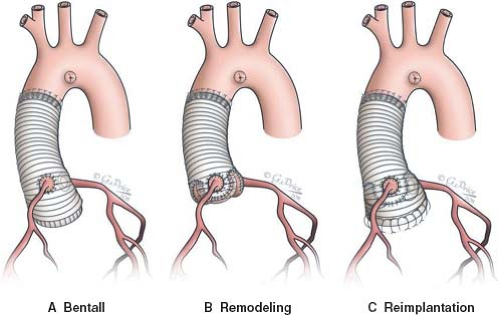 Figure 34.3 A: Bentall procedure, composite graft replacement of the aortic valve, and the ascending aorta. The aortic cusps and sinuses are excised completely. A valved conduit is sutured to the aortic annulus and the coronary arteries are reimplanted into the graft. B: Aortic root replacement with remodeling of the aortic root. A tubular Dacron graft is used with the proximal end tailored to reconstruct the aortic sinuses. The coronary arteries are reimplanted into their respective neoaortic sinuses. C: Aortic root replacement with reimplantation of the aortic valve. The aortic sinuses are excised leaving a trim of aortic tissue attached to the aortic annulus and around the coronary artery orifices. The aortic valve is reimplanted inside a tubular Dacron graft using two suture lines, one below and one above the aortic annulus. The coronary arteries are reimplanted into their respective aortic sinuses and the graft is anastomosed to the distal ascending aorta (52). |
Surgery for aortic root aneurysm in children with MFS has been reported to be safe with good long-term results (50). If the aortic valve is affected, a composite replacement of the valve and ascending aorta is performed. In cases with no substantial aortic valve disease, valve-sparing techniques can be applied, including remodeling of the aortic root or reimplantation of the aortic valve (Fig. 34.3). Creation of neoaortic sinuses in the reimplantation technique is thought to enhance the durability of repair in MFS (51). Current recommendations for elective aortic root surgery in adults with MFS are based on the positive correlation between aortic diameters and risk of dissection. Nonetheless, there is a lack of similar data in children owing to the rarity of aortic complications in this age group. Due to the absence of general consensus in the pediatric population, most centers advise prophylactic surgery using the diameter criterion for adults (∼50 mm) but also take into consideration the presence of accelerated aortic growth (>10 mm/yr), development of aortic regurgitation, or need for mitral valve surgery (37). Following aortic root surgery, close follow-up and continuation of medical therapy is necessary, since distal segments of the aorta may also be affected (40).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


